NADPH Quantification Battle: LC-MS vs. Enzyme Cycling – A Complete Guide for Researchers
This comprehensive analysis compares the two predominant methods for NADPH quantification in biomedical research: Liquid Chromatography-Mass Spectrometry (LC-MS) and Enzyme Cycling Assays.
NADPH Quantification Battle: LC-MS vs. Enzyme Cycling – A Complete Guide for Researchers
Abstract
This comprehensive analysis compares the two predominant methods for NADPH quantification in biomedical research: Liquid Chromatography-Mass Spectrometry (LC-MS) and Enzyme Cycling Assays. The article explores the foundational biochemistry of NADPH, provides detailed methodological protocols, addresses common troubleshooting scenarios, and presents a rigorous, data-driven validation of both techniques. Designed for researchers, scientists, and drug development professionals, this guide synthesizes the latest information to empower informed decision-making in metabolic and redox biology studies, from basic research to therapeutic development.
The Core of Cellular Redox: Understanding NADPH's Role and Measurement Imperative
Within the broader thesis comparing LC-MS and enzyme cycling methods for NADPH quantification, a foundational understanding of NADPH's biochemistry is essential. This cofactor is a central metabolic currency, distinct from its redox counterpart NADH. Its quantification accuracy is critical for research in oxidative stress, anabolism, and xenobiotic metabolism, directly impacting drug development.
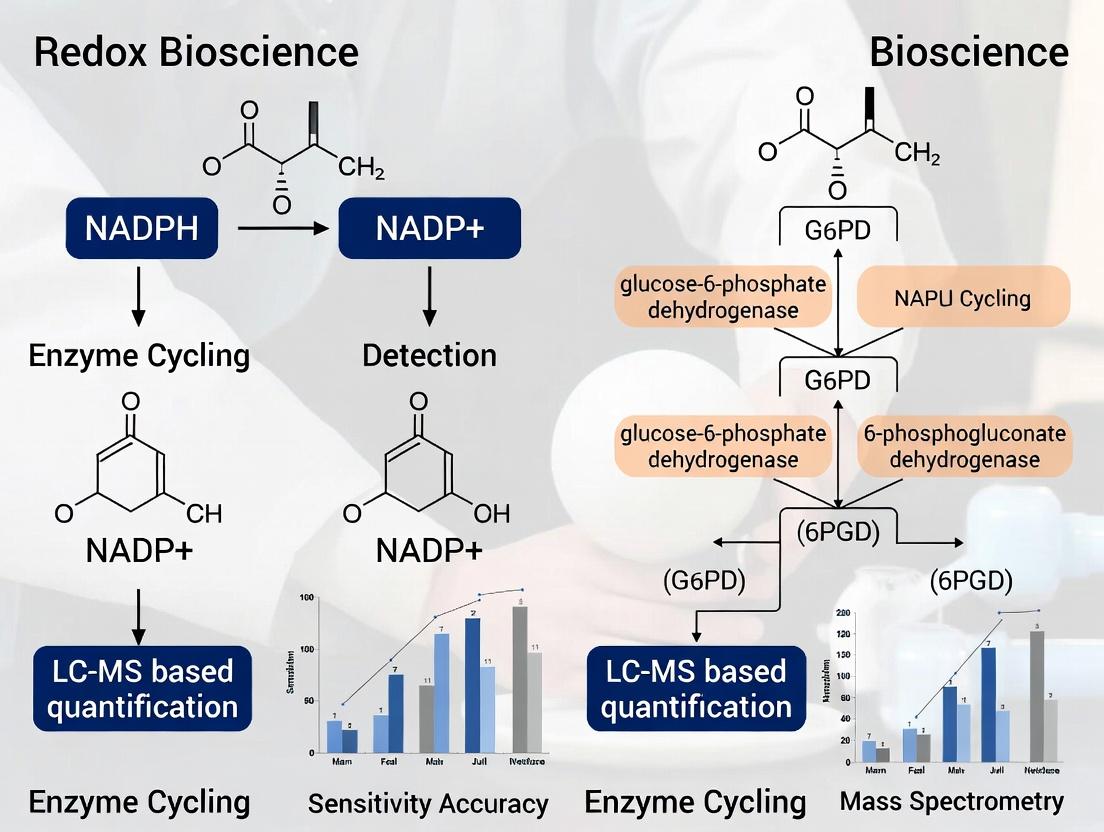
Biochemistry and Metabolic Pathways
NADPH is synthesized primarily through the oxidative branch of the pentose phosphate pathway (PPP), catalyzed by glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase. Other sources include malic enzyme (ME1) and isocitrate dehydrogenase 1 (IDH1) in the cytosol. NADPH's defining feature is its extra phosphate group at the 2' position of the adenine ribose, which creates a binding site for NADPH-specific enzymes, segregating its pool from NADH.
Diagram Title: Primary NADPH Synthesis via Pentose Phosphate Pathway
Critical Cellular Roles
Redox Balance and Antioxidant Defense
NADPH is the essential electron donor for regenerating reduced glutathione (GSH) from oxidized glutathione (GSSG) via glutathione reductase. It also powers the thioredoxin and peroxiredoxin systems. This role is paramount in managing reactive oxygen species (ROS), a key focus in cancer and neurodegenerative disease research.
Diagram Title: NADPH in Antioxidant Defense & Redox Homeostasis
Biosynthetic Reactions
NADPH provides reducing power for anabolism, including fatty acid, cholesterol, and nucleotide synthesis. This makes NADPH metabolism a target in oncology, as proliferating cells have heightened anabolic demands.
Cytochrome P450 Detoxification
In the liver, NADPH is the electron donor for cytochrome P450 oxidoreductase (POR), which fuels Phase I xenobiotic metabolism. Accurate NADPH quantification is thus vital in pharmacokinetic and toxicology studies.
Quantitative Data: NADPH in Biological Systems
Table 1: Typical NADPH Concentrations and Ratios in Mammalian Cells
| System / Cell Type | [NADPH] (approx.) | [NADPH]/[NADP+] | Notes |
|---|---|---|---|
| Hepatocyte (resting) | 50 - 100 µM | ~100:1 to 200:1 | High demand for biosynthesis & detox. Primary measurement context. |
| Erythrocyte | 5 - 15 µM | >100:1 | Sole source is PPP; critical for combating oxidative stress in hemoglobin. |
| Cancer Cell Line (e.g., HeLa) | Variable; often elevated | Can be lower than normal | High flux through PPP; ratio indicative of redox stress. |
| Mitochondrial Matrix | ~10% of cytosolic pool | Similar to cytosol | Sourced via ME2 & IDH2. Important for intramitochondrial antioxidant defense. |
Table 2: Comparison of NADPH Quantification Methodologies
| Parameter | Enzyme Cycling Assay | LC-MS/MS |
|---|---|---|
| Principle | Kinetic measurement of NADPH-dependent reduction of a probe. | Physical separation and detection of NADPH mass. |
| Sensitivity | High (pmol-nmol level) | Very High (fmol-pmol level) |
| Specificity | Moderate (may cross-react with NADH if not optimized) | High (distinguishes NADPH from NADH, NADP+, etc.) |
| Throughput | High (plate-based) | Lower (serial analysis) |
| Sample Preparation | Simpler (protein precipitation often sufficient) | Complex (requires quenching, extraction, sometimes derivatization) |
| Cost per Sample | Low | High |
| Key Advantage in Research | Excellent for rapid, high-throughput screening of many samples. | Gold standard for absolute quantification and redox ratio (NADPH/NADP+). |
Detailed Experimental Protocols
Protocol 1: Enzymatic Cycling Assay for NADPH Quantification (Microplate)
Application Note: This protocol is optimized for specificity towards NADPH over NADH, using glucose-6-phosphate dehydrogenase (G6PD) for high-throughput screening in cell lysates.
Materials:
- Assay Buffer: 50 mM Tris-HCl (pH 8.0), 0.1% BSA.
- Enzyme Solution: 2 U/mL G6PD (from Leuconostoc mesenteroides), 5 mM glucose-6-phosphate (G6P) in assay buffer.
- Detection Probe: 0.2 mM resazurin sodium salt, 5 U/mL diaphorase in assay buffer.
- NADPH Standard: 0-20 µM prepared in assay buffer from a fresh stock.
- Protein Precipitant: 0.5 M perchloric acid (PCA) or 10% trichloroacetic acid (TCA), neutralized with 2 M KOH/0.5 M MOPS.
Procedure:
- Sample Preparation: Lyse 1x10⁶ cells in 100 µL of ice-cold protein precipitant. Vortex and incubate on ice for 5 min. Centrifuge at 16,000 x g for 10 min at 4°C. Transfer supernatant to a fresh tube and neutralize immediately (pH 7-8). Keep on ice.
- Reaction Setup: In a black or clear 96-well plate, add:
- 50 µL of standard or neutralized sample.
- 100 µL of freshly prepared Enzyme Solution.
- 50 µL of Detection Probe.
- Kinetic Measurement: Mix gently and immediately start reading fluorescence (Ex/Em = 540/590 nm for resorufin) or absorbance (A₅₇₀ for resazurin reduction) every minute for 30-60 minutes at 37°C.
- Data Analysis: Calculate the slope (rate) of signal increase for each well. Generate a standard curve from NADPH standards (rate vs. concentration). Interpolate sample concentrations from the linear region of the standard curve.
Protocol 2: LC-MS/MS-Based Quantification of NADPH and NADP+
Application Note: This protocol details a targeted metabolomics approach for absolute quantification of the NADPH/NADP+ ratio, critical for assessing cellular redox state.
Materials:
- Extraction Solvent: 80:20 (v/v) Methanol:Water, chilled to -80°C. Include internal standards (e.g., ¹³C-NADPH, ¹³C-NADP+ if available).
- LC Mobile Phase A: 10 mM Tributylamine, 15 mM acetic acid in water (pH ~5.0).
- LC Mobile Phase B: Methanol.
- LC Column: Reversed-phase (e.g., C18) with ion-pairing compatibility or HILIC column (e.g., Amide).
- MS/MS System: Triple quadrupole operated in negative ion MRM mode.
Procedure:
- Rapid Metabolite Quenching & Extraction: Aspirate media from cultured cells (e.g., in a 6-well plate). Immediately add 1 mL of -80°C extraction solvent directly onto the cells. Scrape cells on dry ice or at -20°C. Transfer extract to a pre-chilled tube.
- Sample Processing: Vortex for 30 sec, then centrifuge at 16,000 x g for 15 min at 4°C. Transfer supernatant to an LC-MS vial. Dry down under nitrogen or vacuum and reconstitute in 50-100 µL of water or starting LC buffer.
- LC-MS/MS Analysis:
- Chromatography: Use ion-pairing or HILIC chromatography to separate NADPH from NADP+ and other nucleotides. A sample gradient: 0-5 min, 0% B; 5-10 min, 0-30% B; 10-12 min, 30-100% B; hold 100% B for 3 min; re-equilibrate.
- MS Detection: Operate in negative electrospray ionization. Use MRM transitions: NADPH (744.1 → 408.1 / 726.1); NADP+ (742.1 → 620.1 / 406.1). Optimize collision energies.
- Quantification: Use calibration curves generated from pure analytical standards spiked into a matrix-matched solution (e.g., extracted from NADPH-depleted cells). Normalize to cell count or protein content.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for NADPH Research
| Reagent / Kit | Function / Application |
|---|---|
| Glucose-6-Phosphate Dehydrogenase (G6PD) | Key enzyme for NADPH-specific enzyme cycling assays. Source (L. mesenteroides) is specific for NADP+. |
| NADPH/NADP+ Fluorometric/Colorimetric Assay Kits | Commercial kits (e.g., from Sigma-Aldrich, Cayman Chemical, Abcam) offer optimized, ready-to-use protocols for high-throughput work. |
| ¹³C/¹⁵N-labeled NADPH Internal Standards | Critical for accurate absolute quantification via LC-MS/MS, correcting for matrix effects and extraction efficiency. |
| Diaphorase & Resazurin | Enzyme/dye couple used in cycling assays to amplify the signal generated by NADPH turnover. |
| Perchloric Acid / Trichloroacetic Acid | Strong acid protein precipitants for metabolite extraction, preserving the labile NADPH pool. |
| Ion-Pairing Reagents (e.g., Tributylamine, DBAA) | Essential for LC-MS separation of highly polar and structurally similar nucleotides like NADPH, NADP+, NADH, NAD+. |
| Siliconized Microtubes | Prevent adsorption of low-concentration NADPH to tube walls during sample preparation. |
Diagram Title: NADPH Quantification Method Selection Workflow
Why Quantify NADPH? Linking Levels to Disease, Drug Response, and Metabolic Phenotype.
NADPH is a critical redox cofactor, serving as the primary electron donor in anabolic biosynthesis (e.g., fatty acids, cholesterol, nucleotides) and for maintaining cellular antioxidant defenses (e.g., via glutathione and thioredoxin systems). Its levels are tightly regulated by a network of enzymes, including glucose-6-phosphate dehydrogenase (G6PD), malic enzyme (ME), and isocitrate dehydrogenase (IDH). Quantifying NADPH is therefore not merely a measurement of a metabolite, but a direct readout of cellular metabolic flux, redox balance, and biosynthetic capacity. Dysregulation of NADPH metabolism is implicated in a wide spectrum of diseases, including cancer, diabetes, neurodegenerative disorders, and rare genetic conditions like G6PD deficiency. In drug development, targeting NADPH-producing pathways (e.g., with G6PD or IDH inhibitors) is a validated strategy, making accurate NADPH quantification essential for assessing target engagement, pharmacodynamics, and therapeutic efficacy.
This document provides detailed application notes and protocols for NADPH quantification, framed within a research thesis comparing the performance characteristics of Liquid Chromatography-Mass Spectrometry (LC-MS) versus Enzyme Cycling Assays.
Comparative Quantitative Data: LC-MS vs. Enzyme Cycling
Table 1: Performance Comparison of NADPH Quantification Methods
| Parameter | LC-MS/MS (Targeted Metabolomics) | Enzyme Cycling (Spectrophotometric) |
|---|---|---|
| Principle | Physical separation and mass-to-charge ratio detection. | Enzymatic reduction of a probe (e.g., MTT, WST-8) coupled to NADPH oxidation. |
| Sensitivity | High (Low pmol to fmol range). | Moderate (High pmol to nmol range). |
| Specificity | Very High. Distinguishes NADPH from NADP⁺, NADH, and other isobars. | Moderate. Can be interfered with by other reducing agents; requires controls. |
| Sample Throughput | Moderate (10-30 min/sample). | High (can be plate-based, <5 min/sample). |
| Sample Requirement | Destructive; requires extraction. | Can be in situ (cell lysates) or extracted. |
| Primary Application | Absolute quantification, isotope tracing, complex biological matrices. | High-throughput screening, kinetic studies in cell lysates. |
| Key Advantage | Multiplexing, superior specificity, gold standard for validation. | Cost-effective, rapid, accessible, no specialized instrumentation. |
| Key Limitation | High instrumentation cost, requires technical expertise. | Indirect measurement, potential for artifactual interference. |
Table 2: Representative NADPH Levels in Biological Systems
| Sample Type | Condition/Treatment | NADPH Level (Approx.) | Method Used | Biological Implication |
|---|---|---|---|---|
| HepG2 Cell Lysate | Control | 20-40 nmol/mg protein | Enzyme Cycling | Baseline redox state. |
| HepG2 Cell Lysate | Treated with 1 μM IDH1 inhibitor (AG-120) | 40-60% decrease | LC-MS/MS | Confirmed on-target drug effect. |
| Patient RBCs | G6PD Deficient (Mediterranean variant) | 5-15% of normal | Enzyme Cycling | Diagnostic for hemolytic anemia risk. |
| Mouse Liver Tissue | High-Fat Diet | 1.5-2.0 fold increase | LC-MS/MS | Adaptation to increased lipogenesis. |
| Cancer Cell Line (AML) | IDH1 R132H Mutant vs. Wild-Type | 2-3 fold increase in 2-HG; Altered NADPH/NADP⁺ ratio | LC-MS/MS | Links oncometabolite production to redox shift. |
Detailed Experimental Protocols
Protocol A: LC-MS/MS-Based Quantification of NADPH from Cultured Cells
Title: Absolute Quantification of NADPH via Hydrophilic Interaction Liquid Chromatography (HILIC) - Tandem Mass Spectrometry.
1. Cell Quenching and Metabolite Extraction:
- Aspirate medium from a 6-well plate (cells at ~80% confluence).
- Rapidly add 1 mL of ice-cold 80% Methanol/Water (v/v, -80°C pre-chilled) to quench metabolism.
- Scrape cells on dry ice and transfer suspension to a pre-chilled microcentrifuge tube.
- Vortex for 30 seconds, then incubate at -80°C for 1 hour.
- Centrifuge at 16,000 x g for 15 minutes at 4°C.
- Transfer supernatant to a fresh tube. Dry under a gentle stream of nitrogen or in a vacuum concentrator.
- Reconstitute the dried extract in 100 μL of HILIC mobile phase A (see below) for LC-MS analysis.
2. LC-MS/MS Analysis:
- Column: SeQuant ZIC-pHILIC (5 μm, 2.1 x 150 mm).
- Mobile Phase A: 20 mM ammonium carbonate, 0.1% ammonium hydroxide in water.
- Mobile Phase B: Acetonitrile.
- Gradient: 85% B to 20% B over 15 min, hold 2 min, re-equilibrate for 8 min. Flow rate: 0.2 mL/min.
- Mass Spectrometer: Triple quadrupole MS in negative electrospray ionization (ESI-) mode.
- MRM Transition for NADPH: 744 > 408 (collision energy optimized). Use stable isotope-labeled NADPH (e.g., ¹³C-NADPH) as an internal standard for absolute quantification.
3. Data Analysis:
- Integrate peak areas for NADPH and its internal standard.
- Generate a calibration curve using pure analytical standards.
- Normalize quantified NADPH amounts to total cellular protein determined from a parallel plate.
Protocol B: Enzyme Cycling Assay for High-Throughput NADPH Quantification
Title: Spectrophotometric NADPH Assay Using Glucose-6-Phosphate Dehydrogenase (G6PD) Cycling.
1. Reagent Preparation:
- Assay Buffer: 100 mM Tris-HCl, pH 8.0.
- Master Mix: Prepare fresh for a 96-well plate. For 1 mL: 800 μL Assay Buffer, 100 μL of 10 mM Glucose-6-Phosphate (G6P), 50 μL of 4 mg/mL MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), 10 μL of 2 U/mL G6PD, 40 μL of 2 mM Phenazine Ethosulfate (PES). Protect from light.
2. Assay Procedure:
- Add 10-20 μL of cell lysate (prepared in neutral PBS) or NADPH standard to a clear 96-well plate.
- Add 100 μL of the Master Mix to each well.
- Immediately start kinetic measurement by reading absorbance at 565 nm (for MTT formazan) every minute for 30-60 minutes at 37°C using a plate reader.
- Critical Control: Include a reaction lacking G6P to account for non-specific reduction of MTT.
3. Calculation:
- Calculate the rate of absorbance increase (ΔA565/min) for each sample and standard.
- Subtract the rate of the no-G6P control.
- Determine NADPH concentration from the linear range of the standard curve (typically 0-200 pmol/well).
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for NADPH Research
| Item | Function & Rationale |
|---|---|
| Stable Isotope-Labeled NADPH (e.g., ¹³C₁₅-NADPH) | Internal standard for LC-MS; enables precise absolute quantification by correcting for matrix effects and extraction losses. |
| Recombinant G6PD Enzyme | Core enzyme for enzyme cycling assays; catalyzes the NADP⁺-dependent oxidation of G6P, generating NADPH. |
| MTT or WST-8 Tetrazolium Salts | Electron acceptors in cycling assays; reduced by PES to form a colored formazan product proportional to NADPH concentration. |
| Phenazine Ethosulfate (PES) | Electron coupling agent; shuttles electrons from NADPH to the tetrazolium salt (MTT/WST-8). |
| ZIC-pHILIC HPLC Column | Stationary phase for polar metabolite separation; critical for resolving NADPH from NADP⁺, NADH, and other nucleotides in LC-MS. |
| NADPH/NADP⁺ Genetically Encoded Biosensor (e.g., iNAP) | For real-time, subcellular resolution monitoring of NADPH:NADP⁺ ratios in live cells via fluorescence microscopy. |
| IDH1/2 Inhibitors (e.g., AG-120/Ivosidenib) | Pharmacological tools to perturb NADPH metabolism in cancer models, linking IDH mutation status to NADPH pool dynamics. |
Visualizing NADPH Metabolism and Measurement Workflows
Diagram Title: NADPH Metabolic Sources, Functions, and Disease Links
Diagram Title: Comparative Workflow: LC-MS vs. Enzyme Cycling for NADPH
This document provides application notes and detailed protocols within the context of a broader research thesis comparing Liquid Chromatography-Mass Spectrometry (LC-MS) and enzymatic cycling assays for the quantification of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) in complex biological matrices. NADPH is a critical redox cofactor involved in biosynthetic pathways and cellular antioxidant defense. Accurate quantification is essential for metabolic studies, drug development targeting metabolic diseases, and oncology research. The primary analytical challenge lies in achieving high sensitivity and specificity amidst interfering compounds in samples like plasma, tissue homogenates, and cell lysates.
Comparative Analytical Performance: LC-MS vs. Enzymatic Cycling
Table 1: Comparative Performance Metrics for NADPH Quantification Methods
| Parameter | Enzymatic Cycling Assay (Spectrophotometric) | LC-MS/MS (Reverse-Phase, ESI-) |
|---|---|---|
| Sensitivity (LLOQ) | ~0.1 µM in well | ~1.0 nM in injection |
| Specificity | Moderate (subject to enzyme specificity) | High (chromatographic separation + MRM) |
| Dynamic Range | 1 - 100 µM | 1 nM - 10 µM |
| Sample Throughput | High (96/384-well plate format) | Moderate (requires chromatographic run time) |
| Sample Volume Required | 10-50 µL | 5-20 µL |
| Matrix Effect Tolerance | Low (highly susceptible to interferents) | Moderate (can be corrected with internal standards) |
| Cost per Sample | Low | High |
| Primary Interferences | Other pyridine nucleotides, sample turbidity | Isobaric compounds, ion suppression |
Detailed Experimental Protocols
Protocol 3.1: Enzymatic Cycling Assay for NADPH Quantification
Principle: NADPH reduces a tetrazolium dye (e.g., WST-8) in a reaction catalyzed by a redox cycling enzyme (e.g., diaphorase), generating a formazan product measured at 450 nm.
Materials:
- Assay Buffer: 100 mM Tris-HCl, pH 8.0.
- Enzyme Solution: Diaphorase (1-5 U/mL in buffer).
- Electron Acceptor: WST-8 (1-2 mM in buffer).
- NADPH Standards: 0, 0.5, 1, 2, 5, 10 µM in assay buffer.
- Biological Sample: Deproteinized cell lysate or plasma (e.g., using perchloric acid precipitation and neutralization).
Procedure:
- Sample Preparation: Deproteinize 50 µL of biological sample with 10 µL of 3M perchloric acid. Incubate on ice for 10 min. Centrifuge at 16,000 x g for 5 min at 4°C. Neutralize supernatant with 5 µL of 3M KOH. Centrifuge again to remove precipitate. Keep samples on ice.
- Reaction Setup: In a 96-well clear plate, combine:
- 80 µL of Assay Buffer.
- 10 µL of standard or processed sample.
- 5 µL of Enzyme Solution.
- 5 µL of Electron Acceptor (WST-8).
- Kinetic Measurement: Mix gently and immediately begin measuring absorbance at 450 nm every 30 seconds for 10-15 minutes using a plate reader maintained at 37°C.
- Data Analysis: Calculate the linear rate (∆A450/min) for each well. Generate a standard curve from rates of NADPH standards. Determine the NADPH concentration in unknowns by interpolation.
Protocol 3.2: LC-MS/MS Quantification of NADPH
Principle: NADPH is separated from matrix components via hydrophilic interaction liquid chromatography (HILIC) and detected using negative electrospray ionization and multiple reaction monitoring (MRM).
Materials:
- Mobile Phase A: 20 mM ammonium acetate in water, pH 9.0 (adjusted with ammonium hydroxide).
- Mobile Phase B: Acetonitrile.
- Internal Standard (IS): ( ^{13}C_{10})-NADPH or NADPH-d4.
- Extraction Solvent: 80:20 Methanol:Water (v/v), chilled to -20°C.
- LC Column: HILIC column (e.g., 2.1 x 100 mm, 1.7 µm particle size).
Procedure:
- Sample Preparation: To 20 µL of biological sample (e.g., cell pellet), add 200 µL of cold extraction solvent spiked with a known amount of Internal Standard. Vortex vigorously for 30 sec. Incubate at -20°C for 20 min. Centrifuge at 18,000 x g for 15 min at 4°C. Transfer supernatant to a fresh tube and dry under a gentle stream of nitrogen. Reconstitute the dried extract in 50 µL of 80% Mobile Phase B / 20% Mobile Phase A.
- LC-MS/MS Conditions:
- Chromatography: HILIC column. Flow rate: 0.3 mL/min. Gradient: 90% B to 50% B over 5 min, hold 1 min, re-equilibrate. Column temp: 30°C.
- MS Detection: ESI negative mode. Source temp: 150°C, desolvation temp: 500°C. MRM transitions:
- NADPH: 744 > 408 (quantifier), 744 > 158 (qualifier).
- IS: Use corresponding transitions for labeled standard.
- Quantification: Generate a calibration curve from analyte/IS peak area ratio vs. known concentration of NADPH standards (1 nM - 10 µM) prepared in a surrogate matrix. Apply this curve to quantify samples.
Visualizations
Title: NADPH Quantification Comparative Workflow
Title: NADPH in Pentose Phosphate & Detection Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials for NADPH Quantification
| Item | Function & Role in Analysis | Example/Catalog Consideration |
|---|---|---|
| Recombinant G6PDH | Key enzyme for enzymatic assay specificity; converts NADP⁺ to NADPH in presence of G6P. | Sigma-Aldrich G7877, recombinant source. |
| Diaphorase (EC 1.8.1.4) | Redox cycling enzyme for signal amplification in enzymatic assays. | Toyobo DIA-301, from Clostridium kluyveri. |
| Tetrazolium Salt (WST-8) | Electron acceptor in cycling assay; reduced to water-soluble formazan for colorimetric detection. | Dojindo 343-07753. |
| Stable Isotope NADPH IS | Internal standard for LC-MS; corrects for matrix effects and variability in extraction. | ( ^{13}C_{10})-NADPH (Cambridge Isotopes). |
| HILIC LC Column | Chromatographic separation of polar NADPH from matrix; critical for MS specificity. | Waters ACQUITY UPLC BEH Amide, 1.7µm. |
| Solid-Phase Extraction (SPE) Cartridges | Optional clean-up for complex matrices (e.g., plasma) prior to LC-MS. | Phenomenex Strata-X-AW (weak anion exchange). |
| NADPH Calibration Standard | High-purity standard for generating quantitative calibration curves in both methods. | MilliporeSigma N1630, ≥97% purity. |
| Perchloric Acid | Effective protein precipitant for sample preparation, preserving labile nucleotides. | ACS grade, suitable for trace analysis. |
Application Notes & Protocols
Core Principles and Application Notes
LC-MS/MS for NADPH Quantification
Principle: Liquid Chromatography coupled with tandem Mass Spectrometry separates NADPH from complex biological matrices via HPLC, followed by detection and quantification based on its unique mass-to-charge ratio (m/z) and fragmentation pattern. It offers high specificity and the ability to multiplex with other metabolites. Key Applications: Direct, absolute quantification of NADPH in tissues (liver, tumor), cells under oxidative stress, and pharmacokinetic studies where drug metabolism alters NADPH pools. Essential for validating enzyme cycling assay results.
Spectrophotometric Enzyme Cycling for NADPH
Principle: An amplified, indirect detection method. NADPH reduces a substrate (e.g., glutathione disulfide, GSSG) in a reaction catalyzed by glutathione reductase (GR). The oxidized glutathione (GSSG) is regenerated in a cyclic manner, leading to the continuous consumption of a colored reagent (e.g., DTNB), measured at 412 nm. The rate of absorbance change is proportional to [NADPH]. Key Applications: High-sensitivity quantification of NADPH in cell lysates, plasma, and mitochondrial fractions where concentrations are low. Ideal for high-throughput screening of compounds affecting NADPH metabolism.
Quantitative Data Comparison
Table 1: Comparative Performance Metrics of NADPH Quantification Methods
| Parameter | LC-MS/MS | Spectrophotometric Enzyme Cycling |
|---|---|---|
| Detection Principle | Physical (Mass, Charge) | Enzymatic Amplification |
| Sample Throughput | Moderate (10-30 samples/run) | High (96-well plate) |
| Sensitivity (LoD) | ~0.1 - 1 nM | ~1 - 10 nM |
| Dynamic Range | 4-5 orders of magnitude | 3-4 orders of magnitude |
| Specificity | Very High (chromatographic separation & MRM) | High (enzyme-specific) |
| Sample Volume | Low (5-50 µL) | Low (10-100 µL) |
| Key Advantage | Multiplexing, absolute quantification | Sensitivity, cost-effectiveness, simplicity |
| Key Limitation | High instrument cost, complex operation | Indirect, susceptible to enzyme inhibitors |
Table 2: Typical NADPH Concentrations in Biological Matrices
| Matrix | Typical Concentration (LC-MS/MS) | Typical Concentration (Enzyme Cycling) |
|---|---|---|
| HepG2 Cell Lysate | 15 - 25 µM | 18 - 30 µM |
| Mouse Liver Tissue | 80 - 120 nmol/g | 75 - 110 nmol/g |
| Human Plasma | 3 - 8 µM | 4 - 10 µM |
| Mitochondrial Fraction | 5 - 15 µM | 8 - 20 µM |
Detailed Experimental Protocols
Protocol A: LC-MS/MS Quantification of NADPH
I. Sample Preparation (Cell Lysate)
- Rapid Quenching: Aspirate media from cultured cells (e.g., in 6-well plate). Immediately add 500 µL of ice-cold 80% Methanol (with 0.1% Formic Acid).
- Extraction: Scrape cells on dry ice. Transfer suspension to a pre-chilled microcentrifuge tube.
- Centrifugation: Spin at 16,000 x g, 4°C for 15 minutes.
- Collection: Transfer supernatant to a new tube. Dry under a gentle stream of nitrogen or in a vacuum concentrator.
- Reconstitution: Reconstitute dried extract in 100 µL of LC-MS grade water. Vortex thoroughly and centrifuge at 16,000 x g for 5 min before LC-MS/MS injection.
II. LC-MS/MS Analysis (Hypothetical Method)
- HPLC System: Reverse-phase C18 column (2.1 x 100 mm, 1.8 µm).
- Mobile Phase: A) 0.1% Formic Acid in Water; B) 0.1% Formic Acid in Acetonitrile.
- Gradient: 0-2 min: 0% B; 2-8 min: 0-30% B; 8-9 min: 30-100% B; 9-11 min: 100% B; 11-12 min: 100-0% B.
- Flow Rate: 0.3 mL/min. Column Temp: 40°C.
- MS/MS: Negative ion mode (ESI-). MRM Transition for NADPH: 744.1 → 408.1 (quantifier) and 744.1 → 272.1 (qualifier). Use stable isotope-labeled NADPH (e.g., ^13C-NADPH) as internal standard.
Protocol B: Spectrophotometric NADPH Enzyme Cycling Assay
I. Reagent Preparation
- Assay Buffer: 100 mM Tris-HCl, 1 mM EDTA, pH 8.0.
- Enzyme Solution: 5 U/mL Glutathione Reductase (GR) in assay buffer (prepare fresh).
- Substrate Solution: 3 mM Glutathione Disulfide (GSSG) in assay buffer.
- Color Developer: 0.5 mM 5,5'-Dithio-bis-(2-nitrobenzoic acid) (DTNB) in assay buffer.
- NADPH Standards: Prepare a dilution series from a stock (e.g., 0, 1, 2, 5, 10, 20 µM) in water.
II. Assay Procedure (96-Well Plate)
- Add Samples/Standards: Pipette 50 µL of standard or deproteinized sample into wells.
- Add Master Mix: Add 150 µL of a freshly prepared master mix containing:
- 135 µL Assay Buffer
- 5 µL GSSG Solution (Final: 75 µM)
- 5 µL DTNB Solution (Final: 25 µM)
- 5 µL GR Solution (Final: 0.25 U/mL)
- Kinetic Measurement: Immediately place plate in a pre-warmed (37°C) plate reader. Monitor absorbance at 412 nm every 30 seconds for 10-15 minutes.
- Calculation: Calculate the linear rate (ΔA412/min) for each well. Plot standard curve (Rate vs. [NADPH]). Determine sample concentration from the curve.
Visualizations
Title: LC-MS/MS NADPH Analysis Workflow
Title: Enzyme Cycling Amplification Principle
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for NADPH Quantification Studies
| Item | Function | Typical Supplier/Example |
|---|---|---|
| NADPH (Standard) | Primary standard for calibration curve generation. | Sigma-Aldrich, Cayman Chemical |
| ^13C-NADPH (Internal Standard) | Isotopically labeled standard for LC-MS/MS to correct for matrix effects & ionization variability. | Cambridge Isotope Laboratories |
| Glutathione Reductase (GR) | Key cycling enzyme for spectrophotometric assay. Critical for assay sensitivity. | Roche, Sigma-Aldrich |
| Glutathione Disulfide (GSSG) | Substrate for GR in the cycling reaction. | Thermo Fisher Scientific |
| 5,5'-Dithio-bis-(2-nitrobenzoic acid) (DTNB) | Colorimetric reagent; reduction yields yellow TNB²⁻ measured at 412 nm. | Sigma-Aldrich |
| Methanol (LC-MS Grade) | For metabolite extraction and LC mobile phase. Ensures low background noise. | Honeywell, Fisher Chemical |
| Formic Acid (LC-MS Grade) | Mobile phase additive to improve ionization efficiency in MS. | Fluka, Thermo Scientific |
| Solid-Phase Extraction (SPE) Cartridges (e.g., SAX) | For sample clean-up to remove interfering ions prior to LC-MS/MS. | Waters, Phenomenex |
Lab Protocols Demystified: Step-by-Step Workflows for LC-MS and Enzyme Cycling
Application Notes and Protocols
1.0 Introduction and Thesis Context Accurate quantification of NADPH is critical in metabolic and cancer research. A core thesis project involves a systematic comparison of Liquid Chromatography-Mass Spectrometry (LC-MS) methods versus traditional enzyme cycling assays for NADPH determination in biological matrices. This protocol details the optimized LC-MS methodology for the quantitative, specific, and sensitive analysis of NADPH, serving as the cornerstone for the comparative analysis.
2.0 Comprehensive Experimental Protocol: LC-MS/MS for NADPH Quantification
2.1 Sample Preparation Workflow Objective: Extract and stabilize NADPH from adherent or suspension mammalian cell cultures (e.g., HEK293, HepG2). Key Principle: Rapid quenching of metabolism and prevention of analyte degradation is paramount. Detailed Protocol:
- Cell Quenching & Lysis: Aspirate culture medium. Rapidly add 1 mL of ice-cold 80% methanol (pre-chilled to -80°C) containing 0.1% formic acid as a stabilizing agent to a 6-well plate well. Immediately place the plate on dry ice for 15 minutes.
- Scraping & Transfer: Scrape cells on dry ice and transfer the slurry to a pre-chilled 1.5 mL microcentrifuge tube.
- Homogenization: Vortex vigorously for 30 seconds. Sonicate on ice using a probe sonicator (3 pulses of 5 seconds at 30% amplitude).
- Protein Precipitation & Clarification: Centrifuge at 21,000 x g for 15 minutes at 4°C. The acidic methanol precipitates proteins.
- Supernatant Collection & Evaporation: Transfer 800 µL of the clear supernatant to a new tube. Dry under a gentle stream of nitrogen gas in a 37°C water bath.
- Reconstitution: Reconstitute the dried metabolite pellet in 100 µL of 10 mM ammonium acetate in water (pH 9.0, adjusted with ammonium hydroxide), optimized for hydrophilic interaction chromatography (HILIC). Vortex for 1 minute.
- Final Clarification: Centrifuge again at 21,000 x g for 10 minutes at 4°C. Transfer 80 µL of the supernatant to a glass insert within an LC-MS vial for analysis. Note: A stable isotope-labeled internal standard (e.g., NADPH-¹³C₁₅N₅) should be added at the lysis step for optimal quantification accuracy.
2.2 Chromatography Parameters (HILIC) Principle: HILIC is ideal for retaining and separating highly polar metabolites like NADPH. System: Agilent 1290 Infinity II LC coupled to 6495C Triple Quadrupole MS. Column: SeQuant ZIC-pHILIC (2.1 x 150 mm, 5 µm, Merck). Mobile Phase:
- A: 20 mM ammonium carbonate + 0.1% ammonium hydroxide in water
- B: Acetonitrile Gradient Program:
| Time (min) | Flow Rate (mL/min) | % B |
|---|---|---|
| 0 | 0.25 | 80 |
| 15 | 0.25 | 20 |
| 17 | 0.25 | 20 |
| 17.5 | 0.25 | 80 |
| 25 | 0.25 | 80 |
Column Temperature: 40°C Autosampler Temperature: 4°C Injection Volume: 5 µL
2.3 Mass Spectrometry Parameters (Negative ESI, MRM) Ion Source: Jet Stream Electrospray Ionization (AJS-ESI), negative mode. Gas & Voltages:
- Drying Gas Temp: 250°C
- Drying Gas Flow: 14 L/min
- Nebulizer Pressure: 30 psi
- Sheath Gas Temp: 400°C
- Sheath Gas Flow: 11 L/min
- Capillary Voltage: 3500 V
- Nozzle Voltage: 500 V Data Acquisition: Dynamic Multiple Reaction Monitoring (dMRM). Optimized MRM Transitions for NADPH:
| Compound | Precursor Ion (m/z) | Product Ion (m/z) | Fragmentor (V) | Collision Energy (V) | Polarity |
|---|---|---|---|---|---|
| NADPH | 742.1 | 158.9 | 380 | 45 | Negative |
| NADPH | 742.1 | 540.0 | 380 | 28 | Negative |
| NADPH-¹³C₁₅N₅ (IS) | 747.1 | 158.9 | 380 | 45 | Negative |
3.0 Quantitative Data Summary for Method Validation Results from a validation run using NADPH spiked into a cell matrix extract.
Table 1: LC-MS/MS Method Performance Characteristics
| Parameter | Result |
|---|---|
| Linear Range | 1 nM – 2000 nM |
| Correlation Coefficient (R²) | 0.9992 |
| Limit of Detection (LOD) | 0.3 nM |
| Limit of Quantification (LOQ) | 1 nM |
| Intra-day Accuracy (% Nominal) | 98.5 - 102.3% |
| Intra-day Precision (% RSD) | 2.1 - 4.8% |
| Inter-day Precision (% RSD) | 3.5 - 6.2% |
| Extraction Recovery (at 100 nM) | 95.4 ± 3.1% |
| Matrix Effect (at 100 nM) | 2.8 ± 1.5% (Suppression) |
4.0 Visualization
4.1 Experiment Workflow
Title: LC-MS Workflow for NADPH Quantification
4.2 Thesis Comparison Framework
Title: Comparative Analysis Framework for NADPH Methods
5.0 The Scientist's Toolkit Table 2: Essential Reagents and Materials for LC-MS NADPH Analysis
| Item (Supplier Example) | Function / Rationale |
|---|---|
| 80% Methanol with 0.1% Formic Acid (in-house prep) | Quenches metabolism, precipitates proteins, and stabilizes labile NADPH via acidification. |
| NADPH (stable isotope-labeled) (Cambridge Isotopes) | Internal Standard (IS) to correct for extraction losses and matrix effects. |
| ZIC-pHILIC LC Column (Merck Millipore) | Stationary phase for HILIC chromatography, enabling retention of polar NADPH. |
| Ammonium Carbonate / Acetonitrile (Optima LC-MS) | High-purity mobile phase components for robust and sensitive MS detection. |
| 0.1% Ammonium Hydroxide (LC-MS Grade) (Fisher) | pH modifier for mobile phase, critical for analyte ionization in negative mode. |
| Filtered Cell Culture Media (Gibco) | Controlled matrix for generating calibration standards in surrogate matrices. |
| Glass LC-MS Vials with Inserts (Agilent) | Prevents leaching of polymers that can cause background interference in the MS. |
This document details application notes and protocols for enzymatic cycling assays used for the ultrasensitive quantification of NADPH. This work is framed within a broader thesis research project comparing the performance of this traditional biochemical method against modern liquid chromatography-mass spectrometry (LC-MS) for NADPH quantification in cellular metabolic studies and drug development screening.
Reagent Composition for NADPH Cycling Assay
The assay amplifies a single NADPH molecule through repeated enzymatic cycles, generating a fluorescent or colorimetric product proportional to the original cofactor concentration.
Table 1: Core Reagent Composition for a Standard NADPH Cycling Assay
| Component | Final Concentration | Function & Notes |
|---|---|---|
| Assay Buffer | 50-100 mM | Tris-HCl or phosphate buffer, pH 8.0. Provides optimal pH for enzymatic activity. |
| NADPH (Sample) | Variable (pM-nM range) | The target analyte for quantification. Heat-inactivated for blank. |
| Glucose-6-Phosphate (G6P) | 1-2 mM | Substrate for G6PDH. Drives the cycling reaction. |
| Glutathione (Oxidized, GSSG) | 0.2-0.5 mM | Electron acceptor for GR, regenerating GSH for the cycle. |
| Glucose-6-Phosphate Dehydrogenase (G6PDH) | 2-5 U/mL | Enzyme 1: Oxidizes G6P, reduces NADP+ to NADPH. |
| Glutathione Reductase (GR) | 2-5 U/mL | Enzyme 2: Oxidizes NADPH to reduce GSSG, completing the cycle. |
| Resazurin | 10-50 µM | Cycling Amplification Reporter. Reduced to fluorescent resorufin by diaphorase (if used) or via non-enzymatic reaction with intermediate electron carriers. |
| Diaphorase | 1-2 U/mL | Optional. Enhances rate of resazurin reduction to resorufin for increased sensitivity. |
| Detergent (e.g., Triton X-100) | 0.1% (v/v) | Prevents enzyme adsorption and maintains stability in low-volume setups. |
Detailed Experimental Protocol
Protocol 1: Setup and Kinetic Measurement of the NADPH Cycling Assay
Objective: To generate a kinetic curve for NADPH detection and determine the linear range of the assay.
Materials:
- Reagents listed in Table 1.
- Black or clear 96- or 384-well microplate.
- Plate reader capable of fluorescence (Ex/Em ~560/590 nm) or absorbance (600 nm) measurement.
- NADPH standard stock solution (e.g., 100 µM in neutral buffer).
Procedure:
- Master Mix Preparation: Prepare a master mix containing all reagents except NADPH (sample/standard) in the assay buffer. Keep on ice.
- Standard Dilution Series: Prepare a serial dilution of NADPH in assay buffer (e.g., 0, 10, 25, 50, 100, 250, 500, 1000 pM). Include a heat-inactivated (80°C for 30 min) NADPH sample as a process blank.
- Plate Setup: Aliquot 80 µL of master mix into each well. Initiate the reaction by adding 20 µL of NADPH standard or unknown sample to respective wells. Perform in triplicate.
- Kinetic Measurement: Immediately place the plate in a pre-warmed (37°C) plate reader. Measure fluorescence/absorbance every 30-60 seconds for 30-60 minutes.
- Data Analysis: Plot the measured signal (Fluorescence Units, FU) against time for each standard concentration. The slope of the linear phase (ΔFU/min) represents the cycling velocity (v).
Protocol 2: Generating a Standard Curve for Quantification
Objective: To create a standard curve relating cycling velocity to NADPH concentration for interpolating unknown samples.
Procedure:
- Follow Protocol 1 to obtain kinetic data.
- For each standard concentration, calculate the mean velocity (v) from the linear portion of the kinetic curve (typically minutes 5-20).
- Subtract the mean velocity of the 0 pM standard (blank) from all other values.
- Plot the blank-corrected velocity (y-axis) against the known NADPH concentration (x-axis).
- Perform linear regression analysis. The resulting equation ([NADPH] = (v - intercept) / slope) is used to calculate concentrations in unknown samples.
Data Presentation and Analysis
Table 2: Representative Kinetic Data and Standard Curve Parameters
| [NADPH] (pM) | Mean Velocity, v (ΔFU/min) | SD | Blank-Corrected v |
|---|---|---|---|
| 0 (Blank) | 15.2 | ± 1.1 | 0.0 |
| 10 | 28.5 | ± 2.3 | 13.3 |
| 25 | 48.1 | ± 3.7 | 32.9 |
| 50 | 82.4 | ± 5.2 | 67.2 |
| 100 | 152.7 | ± 8.9 | 137.5 |
| 250 | 352.0 | ± 15.4 | 336.8 |
| 500 | 655.1 | ± 28.1 | 639.9 |
| 1000 | 1208.5 | ± 45.3 | 1193.3 |
Standard Curve Linear Range: 10 - 1000 pM Regression Equation: y = 1.192x + 1.567 R² Value: 0.9994 Limit of Detection (LOD, 3xSD blank): ~3 pM Limit of Quantification (LOQ, 10xSD blank): ~9 pM
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for NADPH Cycling Assays
| Item | Function/Application | Example Vendor/Product Notes |
|---|---|---|
| Recombinant G6PDH & GR | High-purity, lyophilized enzymes ensure consistent specific activity and low background. | Sigma-Aldrich, Roche. Recombinant forms preferred for lot-to-lot consistency. |
| Ultra-Pure NADPH Standard | Critical for accurate standard curves. Must be quantified and verified (A340 nm, ε=6220 M⁻¹cm⁻¹). | Thermo Fisher, Biomol. Sold as lithium salt, stable at -80°C in dry, pH-neutral aliquots. |
| Resazurin Sodium Salt | Cycling detection reagent. More stable in its sodium salt form. Prepare fresh stock in buffer, protect from light. | Cayman Chemical, Alfa Aesar. |
| 384-Well Low-Volume Assay Plates | Enables high-throughput, reduced reagent consumption for screening applications. | Corning, Greiner. Black plates for fluorescence; clear for absorbance. |
| Non-Adhesive Microplate Sealing Film | Prevents evaporation during kinetic reads, critical for reaction rate consistency. | Thermo Fisher, Excel Scientific. |
| LC-MS Grade Water & Buffers | For reagent preparation to minimize trace contaminants that could affect enzyme kinetics or LC-MS comparison. | Millipore, Honeywell. |
Visualizations
NADPH Enzymatic Cycling Core Reaction Pathway
NADPH Cycling Assay Protocol Workflow
Thesis Context: ECA vs LC-MS Method Comparison
Within a broader thesis comparing LC-MS and enzyme cycling assays for NADPH quantification, sample preparation is the critical foundational step. The accuracy, precision, and reproducibility of both analytical platforms are profoundly influenced by the methods used to generate cell lysates, tissue homogenates, and plasma/serum. This note details optimized protocols for each matrix to ensure the reliable extraction and stabilization of NADPH and related metabolites for downstream comparative analysis.
I. Protocols for Sample Preparation
Protocol 1: NADPH Extraction from Cultured Cell Lysates
Principle: Rapid quenching of metabolism followed by efficient lysis to extract labile NADPH.
- Quenching & Washing: Aspirate culture medium. Immediately add 5 mL of ice-cold PBS (pH 7.4) to the monolayer/adherent cells, swirl, and aspirate. Repeat once.
- Metabolite Extraction: Add an appropriate volume of ice-cold Extraction Buffer (e.g., 70:30 Methanol:Water with 0.1M Formic Acid, or 80% methanol buffered with 10mM ammonium bicarbonate, pH 9.0). Use 500 µL per 1x10⁶ cells.
- Scraping & Transfer: Place the dish/plate on a chilled metal block. Scrape cells quickly and transfer the suspension to a pre-cooled microfuge tube.
- Incubation: Vortex for 10 seconds, then incubate on dry ice or at -80°C for 15 minutes.
- Centrifugation: Centrifuge at 16,000 x g for 15 minutes at 4°C.
- Storage: Transfer the clear supernatant to a new pre-cooled tube. Evaporate to dryness under a gentle nitrogen stream or in a vacuum concentrator. Reconstitute in LC-MS compatible mobile phase or enzyme cycling assay buffer just prior to analysis. Store dried or reconstituted extracts at -80°C.
Protocol 2: NADPH Extraction from Tissue Homogenates
Principle: Mechanical disruption under cryogenic conditions to inhibit degradation.
- Tissue Handling: Snap-freeze tissue in liquid nitrogen immediately after dissection. Weigh frozen tissue on dry ice.
- Pre-homogenization: Place ~20 mg of frozen tissue into a pre-cooled (liquid N₂) mortar or a cryomill tube with a stainless-steel bead.
- Cryogenic Grinding: Pulverize the tissue to a fine powder under continuous liquid nitrogen cooling.
- Metabolite Extraction: Add 500 µL of ice-cold Extraction Buffer (as in Protocol 1, but volume scaled to tissue weight, typically 10-20 µL/mg) to the powder. Homogenize further using a pre-cooled pestle or a tissue lyser (30 Hz, 2 minutes, 4°C).
- Centrifugation: Centrifuge the homogenate at 16,000 x g for 15 minutes at 4°C.
- Clarification: Transfer the supernatant to a clean tube. Perform a second centrifugation if debris persists.
- Storage: Proceed with drying, reconstitution, and storage as in Protocol 1, Step 6.
Protocol 3: Plasma/Serum Preparation for NADPH Analysis
Principle: Rapid separation from cellular components and deproteinization to halt enzymatic activity.
- Blood Collection: Draw blood into tubes containing an appropriate anticoagulant (e.g., K₂EDTA for plasma) or a clot activator (for serum). For NADPH, EDTA is preferred for chelation.
- Immediate Processing: Place tubes on ice and process within 15 minutes.
- Separation: For plasma: Centrifuge at 2,000 x g for 10 minutes at 4°C. For serum: Allow to clot for 30 minutes at RT, then centrifuge at 2,000 x g for 10 minutes at 4°C.
- Aliquoting: Carefully transfer the top plasma/serum layer to a clean tube, avoiding the buffy coat or clot.
- Deproteinization: Mix 50 µL of plasma/serum with 200 µL of ice-cold methanol (or acetonitrile). Vortex vigorously for 60 seconds.
- Centrifugation: Centrifuge at 16,000 x g for 15 minutes at 4°C to pellet proteins.
- Storage: Transfer the clear supernatant to a new tube. Dry down and reconstitute, or directly store at -80°C. Avoid repeated freeze-thaw cycles.
Table 1: Comparison of NADPH Recovery and Stability from Different Matrices Using Optimized Protocols
| Matrix | Optimal Extraction Solvent | Avg. NADPH Recovery (%)* | Key Stability Consideration | Suited for LC-MS? | Suited for Enzyme Assay? |
|---|---|---|---|---|---|
| Cell Lysates | 80% Methanol / 10mM NH₄HCO₃ (pH 9.0) | 95 ± 5 | Rapid quenching is critical; pH 9 reduces degradation. | Excellent | Good (requires pH adjustment) |
| Tissue Homogenates | 70:30 Methanol:Water / 0.1M Formic Acid | 85 ± 8 | Cryogenic pulverization essential for reproducibility. | Excellent | Poor (acidic extract inhibits enzymes) |
| Plasma/Serum | Cold Methanol (4:1 solvent:plasma ratio) | 78 ± 10 | Immediate processing and deproteinization are mandatory. | Good | Fair (matrix effects can interfere) |
*Recovery percentages are estimated relative to a spiked internal standard (¹³C-NADPH) and can vary by cell/tissue type.
III. The Scientist's Toolkit: Essential Reagents & Materials
Table 2: Key Research Reagent Solutions for NADPH Sample Preparation
| Item | Function & Rationale |
|---|---|
| K₂EDTA Blood Collection Tubes | Prevents coagulation for plasma; EDTA chelates metals, slowing NADPH oxidation. |
| Cryogenic Mortar & Pestle or Cryomill | Enables pulverization of frozen tissue without metabolite degradation. |
| Ice-cold Methanol (HPLC Grade) | Universal quenching/extraction agent; denatures enzymes, penetrates cells rapidly. |
| Buffered Extraction Solvents (e.g., NH₄HCO₃ pH 9.0) | Maintains pH to stabilize labile metabolites like NADPH during extraction. |
| ¹³C or ¹⁵N-labeled NADPH Internal Standard | Critical for LC-MS; corrects for losses during preparation and matrix effects. |
| Protein LoBind Microcentrifuge Tubes | Minimizes analyte adhesion to tube walls, improving recovery of low-abundance metabolites. |
| Vacuum Concentrator (with cold trap) | Enables gentle, rapid removal of extraction solvent for sample reconstitution. |
IV. Visualized Workflows & Pathways
Diagram 1: Comparative Sample Prep Workflow for LC-MS vs Enzyme Assay
Diagram 2: NADPH in Pentose Phosphate & Redox Pathways
Within the context of comparative research on LC-MS versus enzyme cycling assays for NADPH quantification, technological optimization is paramount. High-throughput automation and microplate reader optimization are critical for generating robust, reproducible data for method validation. This application note details protocols and configurations essential for modern enzymatic NADPH quantification, enabling researchers to effectively compare its performance against gold-standard LC-MS methodologies.
Key Experimental Protocols
Protocol: Automated High-Throughput NADPH Enzyme Cycling Assay
This protocol is designed for a 384-well format using a liquid handler and an optimized multimode plate reader.
Materials:
- Automation System: Hamilton Microlab STAR or equivalent liquid handling station.
- Microplate Reader: BMG LABTECH CLARIOstar Plus or BioTek Synergy Neo2 with optimized optics.
- Assay Plate: Corning 384-well black-walled, clear-bottom plates (Cat # 3762).
- NADPH Standard: MilliporeSigma, prepared in assay buffer.
Procedure:
- Automated Reagent Dispensing:
- Program liquid handler to dispense 20 µL of NADPH standard (0-100 µM range) or cell lysate sample into designated wells.
- Dispense 30 µL of cycling assay master mix containing: 100 mM Tris-HCl (pH 8.0), 2 mM EDTA, 0.5 mg/mL MTT, 2 mg/mL PES, 5 U/mL G6PD, and 2 mM Glucose-6-Phosphate.
- Kinetic Measurement:
- Immediately transfer plate to pre-warmed microplate reader (30°C).
- Kinetic measurement: Absorbance at 578 nm (MTT formazan) every 30 seconds for 15 minutes.
- Data Processing:
- The automated system exports slope (ΔA/min) values for each well. NADPH concentration is calculated from a standard curve fitted with a 4-parameter logistic (4PL) model.
Protocol: Microplate Reader Optimization for Sensitivity
Maximizing signal-to-noise (S/N) is essential for detecting low NADPH levels in cell lysates.
Procedure:
- Optical Path Calibration:
- Perform a full wavelength scan (500-600 nm) for the 0 and 100 µM NADPH standard to confirm peak absorbance at 578 nm.
- Run a homogeneity test on a fully developed 100 µM standard well to ensure consistent reading across the well.
- Dynamic Range Adjustment:
- Set the reader to "Optimal Gain" or use automatic gain adjustment for the 578 nm filter to prevent signal saturation in the highest standard.
- For fluorescence-based cycling assays (resorufin product, Ex/Em 530-560/580-590), set the bandwidth to 12-15 nm and use a dichroic filter of 570 nm to increase S/N.
- Integration Time:
- Increase the integration time for absorbance measurements to 200-300 ms per read to improve precision for low-concentration samples.
Data Presentation: Comparative Performance Metrics
The following table summarizes quantitative data comparing manual versus automated enzyme cycling assays, benchmarked against LC-MS.
Table 1: Performance Comparison of NADPH Quantification Methods
| Parameter | Manual Enzyme Cycling (96-well) | Automated Enzyme Cycling (384-well) | LC-MS (Reference Method) |
|---|---|---|---|
| Assay Throughput (samples/hour) | 40 | 960 | 20 |
| Assay Volume | 100 µL | 50 µL | 20 µL (post-extraction) |
| Linear Dynamic Range | 0.5 – 50 µM | 0.2 – 100 µM | 0.01 – 100 µM |
| Limit of Detection (LOD) | 0.3 µM | 0.1 µM | 0.005 µM |
| Inter-Assay CV (%) | 8.5% | 4.2% | 3.8% |
| Z'-Factor (Robustness) | 0.65 | 0.82 | N/A |
| Sample Prep Time | 1.5 hours | 1.5 hours (+ 0.5 hr automation setup) | 3 hours (metabolite extraction) |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Automated NADPH Quantification Workflows
| Item | Supplier Example | Function in Workflow |
|---|---|---|
| NADPH Quantification Kit | BioVision (K347-100) / Abcam (ab186031) | Provides optimized, lyophilized master mix for consistent enzyme cycling assay performance. |
| 384-Well Assay Plates | Corning (3762) / Greiner (781098) | Black walls minimize optical crosstalk; clear bottom compatible with microscope validation. |
| Automation-Compatible Tips | Hamilton (235986) / Tecan (10612601) | Low-volume conductive tips for precise, non-contact dispensing of reagents. |
| Plate Reader Validation Kit | BMG LABTECH (UV-Starmap) | Validates absorbance and fluorescence path calibration across the microplate. |
| Cell Lysis Buffer (NADPH stable) | MilliporeSigma (J67302) / Cayman Chemical (10009351) | Inhibits enzymatic degradation of NADPH post-lysis, stabilizing analyte for batch processing. |
| Data Analysis Software | Genedata Screener / IDBS ActivityBase | Enforces automated curve fitting, QC flagging (e.g., +/- 3 SD), and direct comparison to LC-MS data sets. |
Visualized Workflows and Pathways
Diagram Title: Enzyme Cycling Reaction Pathway for Detection
Diagram Title: Automated HTS Workflow from Cells to Data
Solving Real-World Problems: Troubleshooting Guide for NADPH Assay Pitfalls
This application note details protocols for troubleshooting critical LC-MS issues—ion suppression, poor recovery, and instrument drift—within the context of validating a robust LC-MS method for NADPH quantification. This validation serves as a core component of a broader thesis comparing the analytical merits of LC-MS versus traditional enzyme cycling assays for NADPH measurement in drug metabolism studies. The reproducibility and accuracy of the LC-MS platform are paramount for a fair comparative analysis.
Table 1: Common LC-MS Challenges & Impact on NADPH Quantification
| Challenge | Primary Cause | Observed Effect on NADPH Analysis | Typical Magnitude of Error |
|---|---|---|---|
| Ion Suppression | Co-eluting matrix components from biological samples (e.g., salts, phospholipids, metabolites). | Reduced ion signal for NADPH, leading to underestimation of concentration. Can vary between samples. | Signal reduction of 20-80% is common. |
| Poor Recovery | Non-specific binding to vial/column surfaces, incomplete protein precipitation, or compound instability. | Low and inconsistent yield of NADPH from the sample preparation process. | Recovery rates can fall below 60%, increasing variability. |
| Instrument Drift | Gradual contamination of ion source, loss of detector sensitivity, or HPLC pump performance decay. | Systematic change in NADPH response factor over a sequence, compromising accuracy in long batches. | Signal intensity drift of >15% over 24 hours is problematic. |
Detailed Experimental Protocols
Protocol 1: Diagnosing and Mitigating Ion Suppression
Objective: To identify and compensate for matrix effects specific to the sample matrix (e.g., cell lysate, plasma) in NADPH analysis.
Materials & Workflow:
- Post-Column Infusion Test:
- Prepare a constant infusion of a pure NADPH standard (e.g., 100 ng/mL) via a T-connector post-column.
- Inject a blank, processed sample matrix (e.g., protein-precipitated plasma) onto the LC system.
- Monitor the NADPH signal. A dip in the baseline corresponds to the elution of suppressing compounds.
Post-Extraction Spiking Experiment:
- Prepare three sets of samples in replicates (n=6):
- Set A (Neat Solution): NADPH standard in mobile phase.
- Set B (Post-Extraction Spike): Blank matrix taken through the entire sample preparation protocol, then spiked with NADPH.
- Set C (Pre-Extraction Spike): Blank matrix spiked with NADPH before sample preparation.
- Calculate the Matrix Factor (MF):
MF = (Peak Area of Set B / Peak Area of Set A) * 100%. - Calculate the Processed Sample Recovery:
Recovery = (Peak Area of Set C / Peak Area of Set B) * 100%. - An MF <85% or >115% indicates significant ion suppression/enhancement.
- Prepare three sets of samples in replicates (n=6):
Mitigation Strategies:
- Chromatographic Optimization: Alter gradient to separate NADPH from early-eluting matrix interferences.
- Enhanced Sample Cleanup: Incorporate phospholipid removal solid-phase extraction (SPE) plates.
- Use of a Stable Isotope-Labeled Internal Standard (SIL-IS): Spike with ( ^{13}C )- or ( ^{15}N )-labeled NADPH before processing. The IS co-elutes with the analyte and experiences identical suppression, correcting for it.
Diagram 1: Ion Suppression Investigation Workflow
Protocol 2: Investigating Poor Recovery
Objective: To pinpoint the stage of sample preparation where NADPH loss occurs and to improve yield.
Methodology:
- Stage-by-Stage Recovery Assessment:
- Spike blank matrix with NADPH at different stages of the sample preparation workflow (e.g., before protein precipitation, after evaporation, after reconstitution).
- Compare peak areas to a reference standard not subjected to the prior steps.
- Addressing Non-Specific Binding:
- Vial/Container Selection: Use low-binding polypropylene tubes and vials with inserts.
- Additives: Include chelating agents (e.g., 1-2 mM EDTA) to stabilize NADPH, and use silanized glassware.
- Optimizing Protein Precipitation:
- Test different organic solvents (e.g., methanol, acetonitrile) at varying ratios (1:2, 1:3, 1:4 sample:solvent).
- Evaluate temperature (-20°C incubation for 15 min) to improve pellet formation.
- Centrifuge at high speed (≥13,000 g) at 4°C for 15 minutes.
Protocol 3: Monitoring and Correcting Instrument Drift
Objective: To ensure consistent instrument response for NADPH throughout an analytical batch.
Quality Control (QC) Protocol:
- Preparation of QC Samples: Prepare pooled matrix samples spiked with NADPH at Low, Mid, and High concentrations within the calibration curve range.
- Bracketing with QC Samples: Inject QC samples at the beginning of the batch, after every 6-10 experimental samples, and at the end of the batch.
- Drift Assessment: Plot the peak area (or area ratio to IS) of the Mid-Level QC versus injection number.
- Corrective Actions:
- Preventive Maintenance: Regularly clean the ion source and sample introduction pathway as per manufacturer guidelines.
- Signal Normalization: Use the response of the SIL-IS for within-run normalization.
- Advanced Software Correction: Employ batch alignment and signal correction algorithms if available.
Diagram 2: Root Causes & Actions for Instrument Drift
The Scientist's Toolkit
Table 2: Research Reagent Solutions for LC-MS NADPH Analysis
| Item | Function in NADPH LC-MS Analysis |
|---|---|
| Stable Isotope-Labeled NADPH (e.g., NADPH-( ^{13}C _{15})) | Ideal internal standard; corrects for ion suppression, recovery loss, and minor instrument drift. |
| Phospholipid Removal SPE Plates (e.g., HybridSPE-PPT) | Selectively removes phospholipids from biological matrices, a major cause of ion suppression. |
| Low-Binding Polypropylene Microtubes & Vial Inserts | Minimizes non-specific adsorption of the analyte to container walls, improving recovery. |
| HPLC-Grade Methanol & Acetonitrile (with 0.1% Formic Acid) | High-purity solvents for mobile phase and protein precipitation; acid enhances positive ionization. |
| Ammonium Acetate Buffer (e.g., 10mM, pH ~7.0) | Volatile buffer compatible with MS; helps maintain NADPH stability in solution during LC separation. |
| Dedicated LC-MS System Suitability Test Mix | A standard containing compounds covering the m/z range of interest to verify sensitivity and resolution before batch runs. |
Implementing these diagnostic protocols and mitigation strategies ensures the generation of reliable, high-quality quantitative data for NADPH. A robust and stable LC-MS method, validated through these troubleshooting steps, forms the essential foundation for a rigorous and meaningful comparative analysis against enzyme cycling assays in metabolic research.
Within the framework of a comparative thesis investigating LC-MS versus enzymatic cycling for NADPH quantification, robust and reproducible enzyme cycling assays are paramount. This application note details systematic troubleshooting for common pitfalls: non-linear kinetics, high background signal, and enzyme instability, which can critically impact data fidelity in pharmacodynamic studies.
Common Issues & Solutions
Non-Linear Kinetics
Non-linearity in the kinetic readout invalidates quantitation. Primary causes include substrate depletion, enzyme inactivation, or rate-limiting steps from coupled enzymes.
Troubleshooting Protocol:
- Objective: Identify the cause of non-linear reaction progress curves.
- Materials: Microplate reader, pre-warmed assay buffer, substrate stocks, purified enzyme, NADPH standard.
- Procedure:
- Prepare a master mix containing all components except the initiating enzyme.
- Aliquot into wells. Initiate reactions by adding a dilution series of the cycling enzyme (e.g., 1:2, 1:5, 1:10 dilutions).
- Monitor absorbance at 340 nm (for NADPH) kinetically for 30-60 minutes.
- Plot signal vs. time for each enzyme concentration. Linear initial phases indicate appropriate conditions.
- Interpretation: If linearity is only achieved at very low enzyme concentrations, substrate depletion or inhibitor presence is likely. If non-linearity persists, test individual coupling enzymes.
High Background Signal
Elevated background noise reduces assay sensitivity and dynamic range, a critical factor when comparing to low-background LC-MS methods.
Troubleshooting Protocol:
- Objective: Determine the source of high background absorbance or fluorescence.
- Materials: Assay buffer components, spectrophotometer/fluorometer, charcoal, desalting columns.
- Procedure:
- Measure signal from complete assay mixture without the initiating substrate. This defines the "reagent background."
- Systematically omit each component (e.g., Enzyme A, Enzyme B, detection reagent) to identify the contaminant source.
- To remediate, treat the offending reagent: pass enzyme stocks through desalting columns to remove small molecules; use charcoal-treated albumin; or repurify critical substrates.
- Data Analysis: Target a reagent background signal of <5% of the lowest standard's signal.
Enzyme Stability Issues
Loss of cycling enzyme activity during storage or the assay run causes signal drift and poor intra-assay precision.
Stability Assessment Protocol:
- Objective: Quantify the activity loss of enzyme stocks over time and under assay conditions.
- Materials: Enzyme aliquots, storage buffers (with/without stabilizers), water bath.
- Procedure:
- Aliquot the cycling enzyme into different storage conditions: -80°C (control), -20°C, 4°C, and with stabilizers (e.g., 0.1% BSA, 50% glycerol).
- At defined time points (Day 0, 7, 30), thaw an aliquot and measure activity in a standardized cycling assay using a mid-range NADPH standard.
- For in-assay stability, pre-incubate the enzyme at the assay temperature (e.g., 37°C) for 0, 10, 30, 60 minutes before initiating the reaction.
- Interpretation: Calculate percentage activity remaining. >20% loss under assay conditions necessitates reformulation or adjusted addition protocols.
Table 1: Impact of Common Issues on NADPH Quantification Assay Parameters
| Issue | Assay Dynamic Range (µM NADPH) | Signal-to-Background (S/B) at 1 µM | Intra-Assay CV (%) | Correlation vs. LC-MS (R²) |
|---|---|---|---|---|
| Optimal Performance | 0.1 - 10 | 15:1 | <5% | 0.995 |
| Non-Linear Kinetics | 0.5 - 5 | 12:1 | 12% | 0.872 |
| High Background | 1.0 - 10 | 3:1 | 8% | 0.915 |
| Enzyme Instability | 0.2 - 8 | 10:1 | 25% | 0.780 |
Table 2: Efficacy of Stabilizers on Cycling Enzyme Activity Recovery
| Stabilizer Condition | Activity After 30 Days at -20°C | Activity After 1 Hour at 37°C |
|---|---|---|
| No Additive | 65% | 45% |
| 0.1% Bovine Serum Albumin (BSA) | 85% | 70% |
| 50% Glycerol | 95% | 60% |
| 5 mM Dithiothreitol (DTT) | 70% | 50% |
| BSA + Glycerol | 98% | 75% |
Detailed Experimental Protocols
Protocol A: Standardized Enzyme Cycling Assay for NADPH
Principle: NADPH reduces a tetrazolium dye (e.g., WST-8) via an electron carrier in a reaction catalyzed by a cycling enzyme (e.g., diaphorase), generating a colored formazan product proportional to NADPH concentration.
Reagents:
- Assay Buffer: 50 mM Tris-HCl, pH 8.0.
- Cycling Enzyme: Diaphorase (0.5-2 U/mL final).
- Electron Carrier: 1-methoxy-PMS (0.1-0.5 mM final).
- Detection Probe: WST-8 (2 mM final).
- NADPH Standards: 0, 0.1, 0.5, 1, 2, 5, 10 µM in buffer.
Procedure:
- Prepare a master mix containing assay buffer, electron carrier, and detection probe.
- Aliquot 80 µL of master mix per well in a clear 96-well plate.
- Add 10 µL of NADPH standard or unknown sample per well.
- Initiate the reaction by adding 10 µL of cycling enzyme solution.
- Incubate at 37°C, protected from light, and measure absorbance at 450 nm kinetically every minute for 30 minutes.
- Calculate rates from the linear portion of the progress curve (typically minutes 5-15).
Protocol B: Diagnostic Assay for Identifying Rate-Limiting Components
Purpose: To pinpoint which component in a coupled system is causing non-linearity or low signal. Procedure:
- Set up four reaction conditions in parallel:
- Complete System: All components.
- Minus Substrate: No initiating substrate (e.g., no sample NADPH).
- Minus Enzyme A: No first cycling enzyme.
- Minus Enzyme B: No second/coupling enzyme.
- Run the Standardized Enzyme Cycling Assay (Protocol A) for all conditions using a 2 µM NADPH standard.
- Compare the initial reaction rates (V0). The condition omitting the rate-limiting component will show a rate disproportionately lower than expected.
Visualizations
NADPH Enzyme Cycling Reaction Pathway
Enzyme Cycling Assay Troubleshooting Workflow
NADPH Quantification: LC-MS vs Enzyme Cycling Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Robust Enzyme Cycling Assays
| Item | Function | Key Consideration |
|---|---|---|
| High-Purity Cycling Enzymes (e.g., Recombinant Diaphorase) | Catalyzes the redox cycling reaction at the core of the assay. | Use recombinant sources for lower contaminant background. Aliquot and store at -80°C with stabilizers. |
| Tetrazolium Salts (e.g., WST-8, MTT) | Terminal electron acceptors that generate a detectable colored formazan. | WST-8 produces a water-soluble formazan, preferable for homogenous assays. Light-sensitive. |
| Electron Mediators (e.g., 1-methoxy-PMS, Meldola's Blue) | Shuttle electrons from the reduced enzyme to the tetrazolium dye. | Critical for reaction rate. 1-methoxy-PMS offers better stability than PMS. Optimize concentration. |
| Stabilizing Agents (e.g., BSA, Glycerol) | Protect enzyme activity during storage and under assay conditions. | BSA adsorbs contaminants; glycerol prevents cold denaturation. Use in combination. |
| Charcoal-Treated Albumin | Provides protein stabilization without introducing small molecule contaminants. | Essential for reducing high background in ultra-sensitive assays. |
| Desalting Columns (e.g., Zeba Spin) | Rapidly exchange buffers and remove small molecules (e.g., residual ammonium sulfate) from enzyme stocks. | Quick method to improve enzyme stability and reduce background. |
| NADPH Calibration Standards | Provides the primary standard curve for absolute quantitation. | Prepare fresh daily in assay buffer from a certified stock. Critical for correlation with LC-MS data. |
Application Notes
Within a thesis comparing LC-MS and enzyme cycling assays for NADPH quantification, a central finding is the vulnerability of the enzymatic method to matrix effects and NADPH instability. This document details protocols to mitigate these issues, ensuring data comparability. LC-MS offers superior specificity but higher cost and complexity; enzyme cycling provides high sensitivity but requires rigorous sample handling to avoid artifacts from interfering substances and cofactor degradation.
Key Challenges:
- Interfering Substances: Common in biological matrices (e.g., tissue homogenates, serum), these can inhibit enzymes (Glucose-6-phosphate dehydrogenase, G6PD) or react non-specifically, causing inflated or suppressed cycling rates.
- NADPH Instability: NADPH is light-sensitive and prone to degradation by phosphatases and oxidases, leading to underestimation of true concentration.
Optimization Strategy: A sample-specific, tiered approach is required, moving from generic preparation to targeted interference removal.
Experimental Protocols
Protocol 1: Assessment of Matrix Effects on Enzyme Cycling
Purpose: To quantify the inhibition/enhancement effect of a sample matrix on the NADPH cycling reaction.
Materials:
- Sample matrix (e.g., deproteinized cell lysate)
- NADPH standard solution (freshly prepared in 20 mM Tris, pH 8.0)
- Enzyme Cycling Reagent (see Toolkit)
- Microplate reader (capable of reading 340 nm)
Procedure:
- Prepare a calibration curve of NADPH (0, 0.5, 1, 2, 4 µM) in Assay Buffer.
- Prepare an identical calibration curve where each standard is spiked into a constant, dilute amount of the sample matrix (e.g., 10% v/v).
- Add 80 µL of each standard (from steps 1 & 2) to a 96-well plate in duplicate.
- Initiate the reaction by adding 20 µL of pre-mixed Enzyme Cycling Reagent.
- Immediately begin kinetic measurement at 340 nm for 10-15 minutes at 30°C.
- Calculate the reaction rate (ΔA340/min) for each well.
Analysis:
- Plot standard curves (Rate vs. [NADPH]) for both sets.
- Compare slopes. A slope ratio (Matrix slope / Buffer slope) of <0.85 or >1.15 indicates significant matrix interference requiring mitigation (see Protocol 3).
Protocol 2: Evaluation of NADPH Stability Under Storage Conditions
Purpose: To determine optimal handling conditions for sample integrity.
Materials:
- NADPH-spiked sample matrix
- Acidic deproteinization solution (e.g., 0.5 M HClO₄)
- Neutralizing solution (e.g., 2 M KOH, 0.5 M MOPS)
- -80°C freezer, ice bath, bench-top.
Procedure:
- Spike a pooled sample matrix with a known concentration of NADPH. Aliquot into multiple tubes.
- Deproteinize half the aliquots immediately (add acidic solution, incubate on ice 10 min, centrifuge, neutralize supernatant). Keep the other half as native samples.
- Subject aliquots to different conditions:
- A: Processed immediately (T=0 control).
- B: Stored on ice for 2 hours before processing.
- C: Stored at room temperature, in light, for 1 hour before processing.
- D: Stored at -80°C (native) for 1 week before processing.
- Quantify NADPH in all aliquots using both the optimized enzyme cycling assay and LC-MS (reference method).
Analysis:
- Calculate recovery (%) relative to the T=0 LC-MS value.
- Establish maximum permissible handling windows.
Protocol 3: Solid-Phase Extraction (SPE) for Removal of Interfering Substances
Purpose: To clean up samples for enzyme cycling when matrix effects are severe.
Materials:
- Weak anion-exchange (WAX) SPE cartridges (e.g., 10 mg/1 mL)
- Deproteinized, neutralized sample supernatant.
- Wash buffer: 20 mM ammonium acetate, pH 6.0.
- Elution buffer: 20 mM ammonium acetate, pH 9.5, with 0.5 M NaCl.
- Vacuum manifold.
Procedure:
- Condition the WAX cartridge with 1 mL methanol, then 1 mL water.
- Equilibrate with 1 mL of wash buffer (pH 6.0). Do not let the bed dry.
- Load the deproteinized, neutralized sample (adjust pH to ~6.0 if needed).
- Wash with 2 x 1 mL of wash buffer. This step removes acidic interferents.
- Elute NADPH (anionic at pH 9.5) with 2 x 0.5 mL of elution buffer.
- Collect eluate, lyophilize, and reconstitute in Enzyme Cycling Assay Buffer for analysis.
- Assess recovery using NADPH-spiked matrix.
Data Presentation
Table 1: Recovery of NADPH Under Different Handling Conditions
| Sample Type | Storage Condition | Duration | Recovery (Enzyme Cycling) | Recovery (LC-MS) |
|---|---|---|---|---|
| Native Lysate | Room Temp, Light | 1 hour | 62.5% ± 5.2% | 95.1% ± 2.1% |
| Native Lysate | On Ice | 2 hours | 88.3% ± 3.7% | 98.4% ± 1.5% |
| Native Lysate | -80°C | 1 week | 85.0% ± 4.5% | 97.8% ± 1.8% |
| Deproteinized | On Ice | 2 hours | 98.5% ± 2.1% | 99.0% ± 1.2% |
Table 2: Impact of SPE Cleanup on Matrix Effects
| Sample Matrix | Slope (Buffer) | Slope (10% Matrix) | Slope Ratio | Slope (Post-SPE) | Slope Ratio (Post-SPE) |
|---|---|---|---|---|---|
| Liver Homogenate | 0.125 ± 0.005 | 0.092 ± 0.004 | 0.74 | 0.120 ± 0.005 | 0.96 |
| Serum | 0.125 ± 0.005 | 0.140 ± 0.006 | 1.12 | 0.123 ± 0.004 | 0.98 |
Mandatory Visualizations
Optimization Workflow for NADPH Assays
NADPH Enzyme Cycling and Interference
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in NADPH Analysis |
|---|---|
| Glucose-6-Phosphate Dehydrogenase (G6PD) | Key cycling enzyme. Catalyzes the reduction of NADP⁺ to NADPH. Source and lot-specific activity must be verified. |
| Phenazine Methosulfate (PMS) | Electron coupler. Transfers electrons from NADPH to the tetrazolium dye (e.g., MTT). Light-sensitive; requires fresh preparation. |
| Methylthiazolyldiphenyl-tetrazolium (MTT) | Tetrazolium dye. Accepts electrons from PMS to form a colored formazan product, measured at 340 nm. |
| Weak Anion Exchange (WAX) SPE Cartridges | Sample clean-up. Selectively binds NADPH (anionic) at neutral pH, allowing removal of neutral/ cationic interferents and enzyme inhibitors. |
| Perchloric Acid (HClO₄) | Acidic deproteinization agent. Rapidly denatures phosphatases and oxidases to preserve NADPH stability in samples. |
| MOPS Buffering System | Used in neutralization. Provides stable pH post-acid treatment without interfering with the enzyme cycling assay. |
| Stable-Labeled NADPH Internal Standard (¹³C-NADPH) | For LC-MS analysis. Corrects for ionization efficiency shifts and matrix effects, providing the highest accuracy. |
Application Notes
In the context of a thesis comparing LC-MS and enzyme cycling assays for NADPH quantification, a rigorous cost-benefit analysis is critical for method selection in academic and industrial drug development. This analysis extends beyond pure reagent costs to encompass instrument capitalization, technician labor, and sample throughput. NADPH, a key redox cofactor, is measured in metabolic studies, antioxidant research, and drug efficacy testing. The choice of assay impacts budget, timeline, and data quality.
Recent market analyses (2024) indicate a trend towards higher reagent costs for high-purity enzymatic cofactors and stable isotope-labeled internal standards for LC-MS. Conversely, open-access policies for core facility instruments can mitigate capital expenditure. The primary benefit of the LC-MS method is multiplexing capability—simultaneously quantifying NADPH, its oxidized form NADP+, and related metabolites—providing richer data per unit sample. Enzyme cycling assays, while highly specific and sensitive, offer a single-analyte readout but require less specialized training to perform. Technician time investment is notably higher for LC-MS method development and initial calibration but balances out in high-throughput, automated runs.
Protocols
Protocol 1: NADPH Quantification by Enzyme Cycling Assay
Principle: NADPH reduces a tetrazolium dye (e.g., WST-8) in the presence of an electron carrier (1-methoxy PMS), generating a water-soluble formazan dye measured at 450 nm. The rate of increase in absorbance is proportional to NADPH concentration.
Materials:
- Assay Buffer (100 mM Tris-HCl, pH 8.0)
- WST-8 reagent (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium)
- 1-methoxy PMS (1-methoxy-5-methylphenazinium methyl sulfate)
- NADPH standard (0-10 µM range)
- Cell lysate or tissue homogenate sample
- 96-well clear microplate
- Plate reader capable of kinetic measurements at 450 nm
Procedure:
- Prepare the working solution: Mix assay buffer, WST-8 (final 0.2 mM), and 1-methoxy PMS (final 0.02 mM). Protect from light.
- Add 90 µL of working solution to each well of a 96-well plate.
- Add 10 µL of NADPH standard (for calibration curve) or sample to designated wells. Include a blank (10 µL of buffer).
- Immediately place the plate in a pre-warmed (37°C) plate reader.
- Measure absorbance at 450 nm every minute for 30 minutes.
- Calculate the slope (ΔA450/min) for each well. Plot standard curve slopes vs. concentration.
- Determine sample NADPH concentration from the linear regression equation.
Protocol 2: NADPH/NADP+ Quantification by LC-MS/MS
Principle: Metabolites are extracted, separated by hydrophilic interaction liquid chromatography (HILIC), and detected via targeted multiple reaction monitoring (MRM) in negative electrospray ionization mode.
Materials:
- 80% methanol/water (v/v, -80°C, for quenching and extraction)
- Internal Standard: ¹³C-NADPH or d-NADPH
- LC-MS grade solvents: Acetonitrile, Ammonium acetate, Ammonium carbonate
- HILIC column (e.g., BEH Amide, 2.1 x 100 mm, 1.7 µm)
- Triple quadrupole LC-MS/MS system
Procedure:
- Extraction: To 50 µL of cell suspension or homogenate, add 200 µL of ice-cold 80% methanol containing internal standard. Vortex, incubate 10 min at -80°C, centrifuge at 16,000 x g for 15 min at 4°C. Transfer supernatant for analysis.
- LC Conditions:
- Column: BEH Amide
- Mobile Phase A: 20 mM ammonium acetate + 20 mM ammonium carbonate in water (pH ~9.0)
- Mobile Phase B: Acetonitrile
- Gradient: 90% B to 40% B over 5 min, hold 2 min, re-equilibrate.
- Flow rate: 0.4 mL/min. Column temp: 40°C.
- MS/MS Conditions:
- Ionization: ESI-negative
- Capillary voltage: 3.0 kV
- Source temp: 150°C
- Desolvation temp: 500°C
- MRM transitions: NADPH (744.1 > 79.9; 744.1 > 408.0), NADP+ (742.1 > 79.9; 742.1 > 408.0).
- Quantification: Use peak area ratio (analyte/IS) against a calibration curve constructed from authentic standards extracted in parallel.
Data Presentation
Table 1: Comparative Cost-Benefit Analysis for NADPH Quantification Methods
| Parameter | Enzyme Cycling Assay | LC-MS/MS Assay |
|---|---|---|
| Reagent Cost per Sample (USD) | $0.80 - $1.50 | $3.50 - $8.00 (includes IS & columns) |
| Capital Instrument Cost | Plate reader: $15,000 - $50,000 | LC-MS/MS: $250,000 - $500,000 |
| Instrument Access Model | Often in-lab; easy access. | Typically core facility; requires booking. |
| Assay Development Time | 1-2 days (optimization of cycling reagents). | 2-4 weeks (method development & validation). |
| Technician Time per 96 Samples | 4 hours (hands-on + analysis). | 8 hours (extraction, run, data processing). |
| Sample Throughput (per day) | 4-6 plates (384-576 samples). | 2-3 batches (96-144 samples). |
| Data Richness | Single analyte (NADPH). | Multiplexed (NADPH, NADP+, related metabolites). |
| Sensitivity (LOD) | ~10 nM | ~1 nM |
| Key Benefit | Low cost, high throughput, simplicity. | Specificity, multiplexing, dynamic range. |
Diagrams
Diagram Title: Enzyme Cycling Assay Reaction Pathway
Diagram Title: LC-MS/MS NADPH Analysis Workflow
Diagram Title: Assay Selection Logic for Researcher
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for NADPH Quantification
| Item | Function in Experiment | Key Consideration |
|---|---|---|
| WST-8 / 1-methoxy PMS Kit | Forms the core detection chemistry for the enzyme cycling assay; generates measurable color. | Stability of reduced PMS is light-sensitive; requires fresh preparation. |
| Stable Isotope-Labeled NADPH (e.g., ¹³C-NADPH) | Serves as an internal standard for LC-MS to correct for extraction losses and matrix effects. | High cost but essential for accurate quantification; purity is critical. |
| HILIC Chromatography Column (e.g., BEH Amide) | Separates highly polar metabolites like NADPH and NADP+ prior to MS detection. | Requires specific high-pH mobile phases and longer equilibration times than RPLC. |
| Ice-cold Methanol/Water (80:20) | Quenches metabolism instantly and extracts polar metabolites efficiently for LC-MS. | Must be pre-chilled to -80°C for effective quenching; batch preparation ensures consistency. |
| NADPH/NADP+ Calibration Standard Set | Creates the standard curve for absolute quantification in both assay types. | Must be prepared in a matrix mimicking the sample to account for recovery. |
Head-to-Head Data Review: Validating Sensitivity, Accuracy, and Practical Utility
Within the thesis research comparing LC-MS and enzyme cycling assays for NADPH quantification, the accurate characterization of method performance is paramount. This application note details the definitions, calculations, and experimental protocols for determining the Limit of Detection (LOD), Limit of Quantification (LOQ), and Dynamic Range. These parameters are critical for validating both analytical platforms and ensuring reliable data in drug metabolism and pharmacokinetic studies.
Definitions and Calculation Methods
The following table summarizes the core definitions and standard calculation approaches for LOD, LOQ, and Dynamic Range, with particular consideration for LC-MS and enzymatic assay contexts.
Table 1: Core Definitions and Calculation Methods
| Parameter | Definition | Typical Calculation (LC-MS) | Typical Calculation (Enzyme Assay) |
|---|---|---|---|
| Limit of Detection (LOD) | The lowest concentration of analyte that can be reliably distinguished from the background noise. | ( \text{LOD} = 3.3 \times \frac{S{y/x}}{m} ) Where ( S{y/x} ) is the residual standard deviation of the regression line and ( m ) is the slope. | Often based on mean blank signal + 3× standard deviation of the blank (for absorbance/fluorescence). |
| Limit of Quantification (LOQ) | The lowest concentration that can be quantified with acceptable precision (typically ≤20% RSD) and accuracy (80-120%). | ( \text{LOQ} = 10 \times \frac{S_{y/x}}{m} ) | Mean blank signal + 10× standard deviation of the blank; confirmed with precision ≤20% RSD at that level. |
| Dynamic Range | The concentration interval over which the instrument response is linear, from the LOQ to the upper limit of quantification (ULOQ). | From LOQ to ULOQ, where the calibration curve exhibits linearity (R² > 0.99) and accuracy/precision are within ±15%. | From LOQ to ULOQ, maintaining linearity of signal vs. concentration and acceptable precision/accuracy. |
Experimental Protocols
Protocol 3.1: Determining LOD and LOQ for LC-MS NADPH Analysis
Objective: To establish the lowest detectable and quantifiable concentration of NADPH using a validated LC-MS/MS method. Materials: NADPH standard, LC-MS/MS system (e.g., Sciex 6500+), analytical column (e.g., HILIC), mobile phases. Procedure:
- Preparation of Calibration Standards: Prepare a series of NADPH standards in matrix (e.g., cell lysate buffer) at concentrations ranging from expected sub-pM to high nM.
- LC-MS/MS Analysis: Inject each standard in replicate (n=5). Key parameters: MRM transition 744→508; negative ion mode.
- Data Analysis:
- Plot peak area vs. nominal concentration.
- Perform a linear regression on the lower end of the curve (e.g., 5-7 points near the baseline).
- Calculate the residual standard deviation (( S_{y/x} )) of the regression.
- Compute LOD and LOQ using the formulae in Table 1.
- Validation: Prepare samples at the calculated LOD and LOQ concentrations (n=6). For LOQ, confirm the relative standard deviation (RSD) is ≤20% and accuracy is within 80-120%.
Protocol 3.2: Determining LOD and LOQ for Enzyme Cycling NADPH Assay
Objective: To determine the sensitivity of a commercial NADPH cycling assay (e.g., from Promega or Cayman Chemical). Materials: NADPH standard, enzyme cycling assay kit, 96-well plate, fluorescent plate reader. Procedure:
- Blank Determination: Run 8 replicate wells containing only assay buffer and reaction mix (no NADPH). Measure the fluorescence (Ex/Em ~540/590 nm).
- Calibration Curve: Prepare NADPH standards across a wide range (e.g., 0.1 nM to 10 µM) in assay buffer. Run in duplicate.
- Data Analysis:
- Calculate the mean and standard deviation (SD) of the blank signal.
- Preliminary LOD = Mean(blank) + 3×SD(blank); Preliminary LOQ = Mean(blank) + 10×SD(blank).
- From the calibration curve, determine the actual concentrations corresponding to these signal values.
- Confirmation: Test samples spiked at the preliminary LOQ concentration (n=6). The assay passes if the measured RSD is ≤20%.
Protocol 3.3: Establishing Dynamic Range for Comparative Studies
Objective: To define the linear working range for both LC-MS and enzyme assay methods for a direct comparison. Materials: NADPH standard series covering 4-6 orders of magnitude. Procedure:
- Parallel Sample Preparation: Prepare a single, serially diluted set of NADPH standards in a relevant biological matrix (e.g., phosphate buffer with low protein).
- Split-Sample Analysis: Aliquot each standard for analysis by both LC-MS (Protocol 3.1) and the enzyme assay (Protocol 3.2). Perform analyses in triplicate.
- Data Processing & Comparison:
- For each method, plot signal response vs. concentration.
- Perform linear and non-linear regression to identify the linear range (where R² > 0.99 and residuals are randomly distributed).
- The Lower Limit of the Dynamic Range is the method's LOQ.
- The Upper Limit of Quantification (ULOQ) is the highest concentration where accuracy remains within ±15% and precision ≤15% RSD.
- Compile results into a comparison table.
Table 2: Hypothetical Comparison for NADPH Quantification
| Parameter | LC-MS Method | Enzyme Cycling Assay |
|---|---|---|
| LOD | 0.05 nM | 2 nM |
| LOQ | 0.2 nM | 5 nM |
| Dynamic Range | 0.2 nM - 5000 nM (4.4 log) | 5 nM - 1000 nM (2.3 log) |
| Linear Regression (R²) | 0.9992 | 0.9985 |
Visualizations
Title: Workflow for LOD/LOQ Determination in Comparative Analysis
Title: Dynamic Range and Key Analytical Limits
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for NADPH Quantification Studies
| Item | Function & Relevance |
|---|---|
| NADPH (Tetrasodium Salt) High-Purity Standard | Primary reference standard for preparing calibration curves in both LC-MS and enzymatic assays. Essential for accurate LOD/LOQ determination. |
| Stable Isotope-Labeled NADPH (e.g., ¹³C-NADPH) | Internal standard for LC-MS. Corrects for matrix effects and ionization variability, improving precision and accuracy for low-level quantification. |
| Commercial NADP/NADPH-Glo or Cycling Assay Kit | Provides optimized enzymes (e.g., diaphorase) and reagents for selective, amplified detection of NADPH via luminescence/fluorescence. Key for enzymatic LOQ. |
| Mass Spectrometry-Grade Solvents (ACN, MeOH, Water) | Critical for LC-MS mobile phases to minimize background noise and ion suppression, directly impacting LOD. |
| HILIC or Ion-Pairing LC Column | For effective separation and retention of the highly polar, hydrophilic NADPH molecule prior to MS detection. |
| Quenched Cell Lysate Matrix (e.g., with Acid) | Biologically relevant matrix for preparing calibration standards to assess method performance under realistic conditions (matrix effects). |
| 96-/384-Well Assay Plates (Low Fluorescence Binding) | For high-throughput enzymatic assays. Plate quality affects background signal and thus the calculated LOD. |
| Precision Microplate Reader (Fluorescence/Luminescence) | Instrument for detecting the enzymatic cycling reaction output. Sensitivity and dynamic range of the reader influence the overall assay LOD/LOQ. |
This document provides detailed application notes and protocols for assessing accuracy and precision in the quantification of NADPH, a critical cofactor in redox metabolism. This work is a core component of a broader thesis comparing two principal quantification methodologies: Liquid Chromatography-Mass Spectrometry (LC-MS) and enzymatic cycling assays. Establishing robust metrics for accuracy (via spike-and-recovery) and precision (via inter-assay variability) is fundamental to validating the comparative performance of these platforms in biological and pharmaceutical research.
Key Experimental Protocols
Protocol 2.1: NADPH Sample Preparation for LC-MS and Enzymatic Assay
Objective: To standardize initial sample processing for parallel analysis.
- Cell/Tissue Lysate: Homogenize sample in cold 80% methanol/20% PBS (v/v) with 0.1% formic acid for LC-MS, or in specific enzymatic assay extraction buffer (e.g., NADPH/NADP extraction buffer, commercially available). Maintain samples at 4°C.
- Protein Precipitation: Centrifuge at 16,000 x g for 15 minutes at 4°C.
- Supernatant Division: Aliquot the clarified supernatant into two equal parts.
- Part A (for LC-MS): Dry under nitrogen or vacuum. Reconstitute in LC-MS mobile phase A (e.g., 0.1% formic acid in water). Filter through a 0.22 µm PVDF membrane.
- Part B (for Enzymatic Assay): Use directly or dilute with the assay buffer as per kit instructions. Avoid freeze-thaw cycles.
- Storage: Process immediately or store at -80°C.
Protocol 2.2: Spike-and-Recovery Experiment for Accuracy Assessment
Objective: To determine the accuracy of each quantification method by measuring the recovery of a known amount of pure NADPH spiked into a biological matrix.
- Matrix Selection: Use a pooled biological sample (e.g., cell lysate) depleted of endogenous NADPH via charcoal treatment or use a matched, analyte-free matrix.
- Spike Preparation: Prepare a concentrated NADPH standard solution in appropriate solvent. Serial dilute to create spike solutions at three concentrations (Low, Mid, High), targeting final spiked levels within the assay's dynamic range.
- Sample Groups (n=6 per group):
- Group 1 (Unspiked Matrix): Matrix + solvent volume equal to spike.
- Group 2 (Spiked Matrix): Matrix + known concentration of NADPH spike.
- Group 3 (Standard in Buffer): Same NADPH spike concentration in pure assay buffer/LC-MS reconstitution solvent (no matrix).
- Analysis: Quantify NADPH in all samples using the LC-MS and enzymatic assay protocols in parallel.
- Calculation:
- Recovery (%) = [(Measured concentration in Spiked Matrix – Measured concentration in Unspiked Matrix) / Theoretical spike concentration] x 100.
- Theoretical spike concentration is derived from Standard in Buffer.
Protocol 2.3: Inter-Assay Precision (Variability) Experiment
Objective: To determine the precision of each method across different runs, days, and operators.
- Sample Preparation:
- Create three QC samples covering the quantification range: Low QC (near lower limit of quantification, LLOQ), Mid QC (mid-range), and High QC (near upper limit of quantification, ULOQ) in the relevant biological matrix.
- Aliquot and store at -80°C.
- Experimental Design: Analyze each QC sample (n=6 per QC level) in three separate analytical runs performed on different days by different analysts.
- Analysis: Quantify NADPH in all QC samples within each run using the established LC-MS and enzymatic assay methods.
- Calculation:
- Calculate Mean and Standard Deviation (SD) for each QC level within each run (intra-assay precision).
- Calculate Mean and SD for each QC level across all three runs (inter-assay precision).
- Express precision as Coefficient of Variation (%CV) = (SD / Mean) x 100.
Data Presentation
Table 1: Summary of Spike-and-Recovery Data for NADPH Quantification Methods
| Method | Spike Level | Theoretical Conc. (µM) | Mean Measured Conc. (µM) | Mean Recovery (%) | %CV (n=6) | Acceptable Range (80-120%) Met? |
|---|---|---|---|---|---|---|
| LC-MS | Low | 1.0 | 0.95 | 95.0 | 4.2 | Yes |
| Mid | 10.0 | 9.7 | 97.0 | 3.1 | Yes | |
| High | 50.0 | 52.1 | 104.2 | 2.8 | Yes | |
| Enzymatic Assay | Low | 1.0 | 1.25 | 125.0 | 8.5 | No |
| Mid | 10.0 | 10.8 | 108.0 | 5.7 | Yes | |
| High | 50.0 | 47.5 | 95.0 | 4.3 | Yes |
Table 2: Inter-Assay Variability Data for NADPH QC Samples
| Method | QC Level | Nominal Conc. (µM) | Mean Conc. (µM) | SD (µM) | %CV | Total n |
|---|---|---|---|---|---|---|
| LC-MS | Low | 1.0 | 1.02 | 0.06 | 5.9 | 18 |
| Mid | 10.0 | 9.89 | 0.41 | 4.1 | 18 | |
| High | 50.0 | 49.2 | 1.58 | 3.2 | 18 | |
| Enzymatic Assay | Low | 1.0 | 1.15 | 0.12 | 10.4 | 18 |
| Mid | 10.0 | 10.5 | 0.84 | 8.0 | 18 | |
| High | 50.0 | 48.1 | 3.21 | 6.7 | 18 |
Visualization Diagrams
Title: Workflow for Comparative NADPH Method Validation
Title: Enzymatic Cycling Assay Core Reaction Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for NADPH Quantification Studies
| Item | Function & Relevance | Example/Note |
|---|---|---|
| NADPH Standard (High Purity) | Primary standard for calibration curves, spike solutions, and accuracy determination. Essential for both LC-MS and enzymatic methods. | Purchase certified reference material. Store aliquoted at -80°C in inert buffer, protected from light. |
| NADPH/NADP Extraction Buffer | Specialized buffer for stabilizing labile pyridine nucleotides during sample homogenization for enzymatic assays. Prevents degradation. | Often contains detergents, chelators, and cycling assay inhibitors. Commercial kits available. |
| Enzymatic NADPH Detection Kit | Provides optimized reagents (enzymes, substrates, probes) for the coupled cycling reaction, ensuring sensitivity and specificity. | Kits from suppliers like Sigma-Aldrich, Cayman Chemical, or Abcam. Includes G6PD, GR, and DTNB. |
| LC-MS Grade Solvents & Additives | High-purity methanol, acetonitrile, water, and formic acid for mobile phases and sample reconstitution. Critical for low background and consistent ionization. | Use solvents specifically labeled for LC-MS to avoid contaminants that suppress ionization. |
| Stable Isotope-Labeled NADPH Internal Standard (e.g., ¹³C-NADPH) | Added to samples prior to LC-MS processing to correct for matrix effects, recovery losses, and instrument variability. Gold standard for quantitative LC-MS. | Can be sourced from Cambridge Isotope Laboratories or C/D/N Isotopes. |
| Charcoal (Activated) | Used to prepare analyte-depleted matrix for spike-and-recovery experiments by adsorbing endogenous NADPH. | Must be thoroughly removed via centrifugation and filtration post-treatment. |
| Quality Control (QC) Pool Matrix | A homogeneous, large-volume pool of the biological matrix of interest (e.g., liver homogenate). Used to prepare QC samples for precision studies. | Characterize its endogenous NADPH level. Aliquot and store long-term at -80°C. |
Application Note 1: Targeting NADPH Metabolism in Glioblastoma
Thesis Context: This study exemplifies the need for robust, precise NADPH quantification to validate LC-MS metabolomics data. Enzyme cycling assays were employed as a secondary validation method to confirm elevated NADPH/NADP+ ratios in IDH1-mutant glioblastoma cells, a key finding with therapeutic implications.
Background: Mutations in Isocitrate Dehydrogenase 1 (IDH1) are oncogenic drivers in glioblastoma and other gliomas. The mutant enzyme (IDH1 R132H) catalyzes the reduction of α-KG to the oncometabolite D-2-hydroxyglutarate (2-HG), consuming NADPH in the process. Paradoxically, cells compensate by upregulating the oxidative pentose phosphate pathway (PPP), leading to a net increase in NADPH, which supports antioxidant defense and biomass production.
Key Quantitative Data:
Table 1: Metabolomic and NADPH Analysis in IDH1-WT vs. IDH1-Mutant Glioblastoma Stem Cells (GSCs)
| Metric | IDH1-Wild Type GSCs | IDH1-Mutant GSCs (R132H) | Measurement Method | Significance (p-value) |
|---|---|---|---|---|
| Intracellular 2-HG | 0.05 ± 0.01 µmol/g protein | 35.2 ± 4.8 µmol/g protein | LC-MS/MS | < 0.001 |
| NADPH Level | 45.3 ± 5.1 pmol/10^6 cells | 82.7 ± 7.6 pmol/10^6 cells | LC-MS | < 0.01 |
| NADP+ Level | 38.9 ± 4.3 pmol/10^6 cells | 25.4 ± 3.1 pmol/10^6 cells | LC-MS | < 0.05 |
| NADPH/NADP+ Ratio | 1.16 ± 0.15 | 3.26 ± 0.41 | Calculated from LC-MS | < 0.001 |
| NADPH/NADP+ Ratio (Validation) | 1.21 ± 0.18 | 3.19 ± 0.38 | Enzyme Cycling Assay | < 0.001 |
| Glucose-6-P Dehydrogenase Activity | 100% ± 8% (Reference) | 215% ± 22% | Spectrophotometric Assay | < 0.001 |
Protocol: Validation of NADPH/NADP+ Ratio via Enzyme Cycling Assay
- Cell Lysis and Extraction: Pellet 1x10^6 cells. Resuspend in 200 µL of cold NADP+/NADPH extraction buffer (0.1N NaOH/1% DTAB for NADPH; 0.1N HCl for total NADP). Vortex vigorously and incubate on ice for 10 min. Centrifuge at 12,000g for 10 min at 4°C. Transfer supernatant to a new tube. Neutralize HCl extracts with NaOH/TRIS; neutralize NaOH extracts with HCl.
- NADPH Detection Reaction: In a 96-well plate, combine:
- 50 µL sample or standard (0-10 pmol NADPH)
- 100 µL Reaction Mix: 100 mM TRIS-HCl (pH 8.0), 0.5 mM MTT, 2 mM PMS, 5 mM G6P, 1 U/mL G6PD.
- Incubation and Measurement: Incubate plate at 37°C for 30-60 minutes, protected from light. Measure absorbance at 565 nm (for MTT formazan) using a plate reader.
- NADP+ Detection: Determine NADP+ by difference. Measure total NADP (NADPH + NADP+) from the acid extract. Subtract the NADPH value (from basic extract) from the total NADP value.
- Calculation: Generate a standard curve from known NADPH amounts. Calculate sample concentrations and compute the NADPH/NADP+ ratio.
Signaling Pathway Diagram:
Diagram 1: NADPH Metabolism in IDH1-Mutant Glioblastoma.
Research Reagent Solutions:
- NADP/NADPH Extraction Buffer (Alkaline/Acid): Selective stabilization of redox pairs for accurate species-specific measurement.
- Glucose-6-Phosphate Dehydrogenase (G6PD): Key enzyme for the cycling assay, catalyzes NADP+ reduction using G6P.
- MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) & PMS (Phenazine methosulfate): Colorimetric detection pair; MTT is reduced to purple formazan by NADPH via PMS.
- D-2-Hydroxyglutarate (2-HG) ELISA Kit: Validates mutant IDH1 activity and oncometabolite production.
- IDH1 R132H Inhibitor (e.g., Ivosidenib): Tool compound to probe metabolic vulnerability and validate target engagement by reversing the metabolic phenotype.
Application Note 2: Monitoring NADPH Decline in Parkinson's Disease Neuronal Models
Thesis Context: This case highlights the sensitivity requirement for NADPH quantification in neurodegeneration research. LC-MS provided absolute quantitation of declining NADPH pools in dopaminergic neurons, while cycling assays offered a high-throughput platform for screening neuroprotective compounds.
Background: Parkinson's disease (PD) involves mitochondrial dysfunction and oxidative stress. NADPH is critical for maintaining glutathione (GSH) in its reduced, antioxidant state. PINK1/Parkin mutations impair mitophagy, leading to ROS accumulation and NADPH depletion, creating a vicious cycle of metabolic and oxidative stress.
Key Quantitative Data:
Table 2: Metabolic Parameters in PINK1-KO vs. Wild-Type Dopaminergic Neurons (LUHMES Cell Line)
| Metric | Wild-Type Neurons | PINK1-Knockout Neurons | Measurement Method | Notes |
|---|---|---|---|---|
| NADPH Level | 62.5 ± 6.8 pmol/10^6 cells | 28.4 ± 5.2 pmol/10^6 cells | LC-MS (Primary) | 54% reduction (p<0.005) |
| GSH/GSSG Ratio | 25.4 ± 3.1 | 8.7 ± 1.9 | Enzymatic Recycling Assay | 66% reduction (p<0.001) |
| Basal ROS Level | 100% ± 12% | 245% ± 31% | DCFDA Fluorescence | (p<0.001) |
| Mitochondrial Membrane Potential (ΔΨm) | 100% ± 9% | 67% ± 11% | TMRE Fluorescence | (p<0.01) |
| Viability after Oxidant (H2O2) | 85% ± 7% | 42% ± 9% | Calcein-AM Assay | (p<0.001) |
| NADPH Rescue (NAC Treatment) | - | 45.1 ± 6.3 pmol/10^6 cells | Enzyme Cycling (HTS) | p<0.05 vs. untreated KO |
Protocol: High-Throughput NADPH Screening for Neuroprotectants
- Cell Culture and Treatment: Plate PINK1-KO LUHMES neurons in 384-well plates. Differentiate for 6 days. Treat with compound library (e.g., N-acetylcysteine (NAC), PPARγ agonists) for 48 hours.
- One-Step NADPH Extraction/Detection: Remove media. Add 20 µL of ice-cold cycling assay lysis/detection buffer directly to wells: 100 mM TRIS (pH 8.0), 0.1% DTAB, 0.5 mg/mL MTT, 0.1 mg/mL PMS, 2 mM G6P, 1 U/mL G6PD.
- Rapid Kinetic Measurement: Immediately place plate in a kinetic plate reader at 37°C. Monitor absorbance at 565 nm every 30 seconds for 15 minutes. The initial rate of absorbance increase is proportional to NADPH concentration.
- Data Analysis: Use the slope (ΔA565/min) from the linear phase (minutes 2-8) for analysis. Compare slopes of treated wells to vehicle and wild-type controls. Z'-factor should be >0.5 for a robust screen.
Metabolic Stress Pathway Diagram:
Diagram 2: NADPH Depletion in Parkinson's Disease Pathway.
Research Reagent Solutions:
- PINK1-Knockout LUHMES Cell Line: Genetically engineered, inducible human dopaminergic neuron model for PD.
- TMRE (Tetramethylrhodamine ethyl ester): Fluorescent potentiometric dye for measuring mitochondrial membrane potential (ΔΨm).
- DCFDA (2',7'-Dichlorofluorescin diacetate): Cell-permeable probe that becomes fluorescent upon oxidation by intracellular ROS.
- Glutathione Assay Kit (Colorimetric/Fluorometric): For simultaneous measurement of GSH and GSSG levels.
- 384-Well Plate-Compatible NADPH Cycling Assay Reagent: Optimized for single-addition, kinetic readout in high-throughput screening formats.
Application Note 3: Elucidating the Mechanism of a Novel NAMPT Inhibitor (KPT-9274) in AML
Thesis Context: This drug mechanism study underscores the importance of paired analytical techniques. LC-MS tracked global metabolomic changes, while enzyme cycling assays provided rapid, specific validation of NADPH depletion as a key pharmacodynamic biomarker in patient-derived samples.
Background: Nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme in the NAD+ salvage pathway. KPT-9274 is a dual NAMPT/PAK4 inhibitor. Inhibition of NAMPT depletes cellular NAD+, which subsequently impairs NADPH production through metabolic coupling, inducing oxidative stress and cell death in acute myeloid leukemia (AML).
Key Quantitative Data:
Table 3: Metabolic Impact of NAMPT Inhibition (KPT-9274) in MV4-11 AML Cells
| Metric | DMSO Control | KPT-9274 (100 nM, 24h) | Measurement Method | Implication |
|---|---|---|---|---|
| Intracellular NAD+ | 450 ± 32 µM | 85 ± 12 µM | LC-MS/MS | 81% depletion (Target Engagement) |
| Intracellular NADPH | 58 ± 7 µM | 19 ± 4 µM | Enzyme Cycling (Validation) | 67% depletion |
| NADPH/NADP+ Ratio | 4.2 ± 0.5 | 1.3 ± 0.3 | LC-MS | Loss of Redox Capacity |
| Lactate/Pyruvate Ratio | 15 ± 2 | 38 ± 5 | LC-MS | Shift toward glycolysis |
| ATP Level | 100% ± 8% | 62% ± 9% | Luminescence Assay | Bioenergetic stress |
| Apoptosis (Annexin V+) | 5% ± 2% | 48% ± 7% | Flow Cytometry | Cell death induction |
| IC50 (72h) | - | 45 nM | Cell Viability (CTB) | Potency |
Protocol: Integrated LC-MS and Enzymatic Validation for Drug Studies
Sample Preparation for Multi-Platform Analysis:
- Treat cells (e.g., AML cell lines or primary blasts) with compound or vehicle.
- Quench metabolism with dry ice/80% methanol (-80°C). Scrape cells and centrifuge.
- Split Extract: Use 80% of supernatant for LC-MS polar metabolite profiling. Use 20% for enzymatic validation assays after drying and reconstituting in appropriate buffer.
LC-MS Metabolomics Protocol:
- Column: HILIC (e.g., BEH Amide, 2.1 x 150 mm, 1.7 µm).
- Mobile Phase: A = 95:5 Water:ACN w/ 20mM Ammonium Acetate (pH 9.0); B = ACN.
- Gradient: 95% B to 40% B over 15 min. Flow: 0.25 mL/min.
- MS: High-resolution mass spectrometer (e.g., Q-TOF) in negative ion mode for NADP(H), nucleotides.
Enzymatic Validation from Reconstituted Extract:
- Follow the NADPH enzyme cycling assay protocol (See App Note 1, Steps 2-5) using the reconstituted extract.
- Correlate NADPH values from LC-MS (absolute quantitation via internal standard) with values from the enzymatic assay (activity-based quantitation).
Drug Mechanism Workflow Diagram:
Diagram 3: Mechanism of NAMPT Inhibitor KPT-9274 in AML.
Research Reagent Solutions:
- KPT-9274 (or other NAMPT Inhibitor, e.g., FK866): Tool compound to induce NAD+ depletion and study downstream metabolic effects.
- NAD/NADH Quantitation Kit (Fluorometric): Complementary to NADP(H) measurement for full pyridine nucleotide analysis.
- HILIC Columns & Stable Isotope-Labeled Internal Standards (e.g., 13C-NAD+, 15N-NADPH): Essential for robust, quantitative LC-MS metabolomics of polar metabolites.
- Annexin V-FITC / Propidium Iodide Apoptosis Kit: To link metabolic changes (NADPH depletion) to phenotypic outcome (cell death).
- CellTiter-Blue (CTB) Cell Viability Assay: A resazurin-based assay whose signal is dependent on NAD(P)H, providing a functional readout of total pyridine nucleotide status.
Introduction Within the broader thesis comparing LC-MS and enzyme cycling for NADPH quantification, selecting the appropriate method is paramount. This decision directly impacts data reliability, throughput, and resource allocation in drug development. This document provides a structured matrix and detailed protocols to guide researchers in making a definitive methodological choice.
Definitive Decision Matrix for NADPH Quantification The following table synthesizes key decision criteria based on research objectives and available resources.
Table 1: Method Selection Decision Matrix
| Criterion | LC-MS/MS (Targeted Quantification) | Enzyme Cycling (Spectrophotometric/Fluorometric) | Primary Consideration |
|---|---|---|---|
| Primary Research Question | Absolute quantification; Specific isomer detection (e.g., NADPH vs. NADP); Stable isotope tracer studies. | Relative quantification of total NADPH/NADP+ ratio; High-throughput screening of metabolic effectors. | Specificity vs. Throughput |
| Sensitivity (Limit of Detection) | ~0.1 - 1 nM (Excellent) | ~0.1 - 1 µM (Good) | Trace-level analysis required? |
| Specificity | Very High (Chromatographic separation + mass detection) | Moderate (Subject to interference from sample matrix) | Sample complexity (e.g., crude lysates) |
| Sample Throughput | Low to Moderate (5-15 min/sample) | Very High (96/384-well plate, <1 min/sample) | Number of samples per study |
| Capital & Recurrent Cost | Very High (instrument, maintenance, skilled operator) | Low (plate reader, standard lab equipment) | Budget and infrastructure |
| Technical Expertise Required | High (method development, data interpretation) | Low to Moderate (routine biochemical assay) | Available personnel skills |
| Sample Destructive? | Yes (typically) | Yes | Need for sample recovery? |
| Key Data Output | Absolute concentration (pmol/mg protein), isotope enrichment ratio. | Relative absorbance/fluorescence units, calculated ratio. | Data reporting requirements |
Detailed Application Notes & Protocols
Protocol 1: NADPH Quantification by LC-MS/MS This protocol is optimized for the separation and detection of NADPH and NADP+ from cell lysates.
I. Reagent Solutions & Sample Preparation
- Extraction Buffer: 80% HPLC-grade methanol, 20% Milli-Q water, with 0.1M ammonium acetate (pH 7.0). Kept at -20°C. Function: Rapid quenching of metabolism and protein precipitation.
- Internal Standard (IS) Solution: Stable isotope-labeled NADPH (e.g., ( ^{13}C_5 )-NADPH) at 100 nM in extraction buffer. Function: Corrects for matrix effects and extraction efficiency losses.
- LC Mobile Phase A: 5 mM ammonium acetate in water, pH 8.0 (adjust with ammonium hydroxide). Function: Aqueous buffer for hydrophilic interaction chromatography (HILIC).
- LC Mobile Phase B: Acetonitrile with 5 mM ammonium acetate. Function: Organic phase for HILIC separation.
II. Experimental Workflow
- Metabolite Extraction: Aspirate medium from cultured cells (e.g., in a 6-well plate). Immediately add 500 µL of cold Extraction Buffer spiked with IS. Scrape cells, transfer to a microtube, vortex for 10s, and incubate at -20°C for 1 hour.
- Protein Precipitation: Centrifuge at 16,000 x g for 15 min at 4°C.
- Sample Clarification: Transfer 400 µL of supernatant to a new tube. Dry under a gentle stream of nitrogen at 30°C.
- Reconstitution: Reconstitute the dried pellet in 100 µL of 80% Mobile Phase B / 20% Mobile Phase A. Vortex thoroughly and centrifuge at 16,000 x g for 10 min at 4°C.
- LC-MS/MS Analysis: Inject 5-10 µL onto the HILIC column.
III. Instrument Parameters (Example)
- Column: BEH Amide HILIC (2.1 x 100 mm, 1.7 µm).
- Gradient: 90% B to 40% B over 8 min, hold 2 min, re-equilibrate.
- Flow Rate: 0.3 mL/min. Column Temp: 40°C.
- MS: Negative ion mode ESI. MRM Transitions: NADPH: 744→408; NADP+: 742→406; ( ^{13}C_5 )-NADPH: 749→413.
Protocol 2: NADPH Quantification by Enzyme Cycling Assay This protocol is adapted for a 96-well plate format to measure total NADPH + NADP+ and NADP+ alone.
I. Reagent Solutions
- NADPH Extraction Buffer: 0.1M NaOH, 0.1% DTAB. Function: Alkaline lysis, stabilizes NADPH by inactivating enzymes.
- NADP+ Extraction Buffer: 0.1M HCl. Function: Acid lysis, converts NADPH to NADP+ for total cofactor measurement.
- Assay Buffer: 100 mM Tris-HCl (pH 8.0), 2 mM EDTA, 0.5 mM MTT, 2 mM PMS, 1 mM G6P. Function: Provides optimal pH and components for enzymatic reaction.
- Enzyme Solution: 4 U/mL G6PDH in Assay Buffer (prepared fresh). Function: Catalyzes the cycling reaction, reducing NADP+ to NADPH.
II. Experimental Workflow
- Sample Preparation (Dual Extraction):
- For NADPH: Add 100 µL of cold NaOH/DTAB buffer to cell pellet, incubate 10 min at 60°C, then neutralize with 100 µL of 0.1M HCl.
- For Total NADP(H): Add 100 µL of cold HCl buffer to a parallel pellet, incubate 10 min at 60°C, then neutralize with 100 µL of 0.1M NaOH/DTAB buffer.
- Centrifuge all extracts at 10,000 x g for 5 min. Use supernatant immediately.
- Plate Setup: In a clear 96-well plate, add 50 µL of sample or NADPH standard (0-10 µM range) per well.
- Reaction Initiation: Add 150 µL of the combined Assay Buffer + Enzyme Solution to each well.
- Kinetic Measurement: Immediately place plate in a plate reader pre-warmed to 37°C. Monitor absorbance at 570 nm every 30 seconds for 10-15 minutes.
- Calculations: Use the linear portion of the absorbance increase (∆A/min). NADPH concentration is derived from the NADPH extract. Total NADP(H) is from the HCl extract. NADP+ = Total - NADPH.
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in NADPH Quantification |
|---|---|
| Stable Isotope-Labeled NADPH (e.g., ( ^{13}C_5 )) | Internal standard for LC-MS; enables correction for matrix effects and absolute quantification. |
| HILIC Chromatography Column (e.g., BEH Amide) | Separates polar metabolites like NADPH and NADP+; critical for specificity in LC-MS. |
| Glucose-6-Phosphate Dehydrogenase (G6PDH) | Key cycling enzyme for spectrophotometric assay; reduces NADP+ while oxidizing G6P. |
| Tetrazolium Dye (MTT or WST-1) | Electron acceptor in enzyme cycling assay; reduced by PMS to form a colored formazan product. |
| Phenazine Methosulfate (PMS) | Electron carrier in cycling assay; shuttles electrons from reduced NADPH to the tetrazolium dye. |
| Dodecyltrimethylammonium Bromide (DTAB) | Detergent in alkaline extraction; helps stabilize NADPH by denaturing degrading enzymes. |
| Ammonium Acetate (HPLC-grade) | Volatile buffer salt for LC-MS mobile phases; promotes ionization and improves chromatographic peaks. |
Pathway and Workflow Visualizations
LC-MS/MS NADPH Quantification Workflow
Enzyme Cycling Assay Reaction Pathway
Method Selection Logic Tree
Conclusion
The choice between LC-MS and enzyme cycling for NADPH quantification is not a matter of declaring a universal winner, but of strategically matching method capabilities to specific research needs. LC-MS offers unparalleled specificity, multiplexing potential, and suitability for complex or discovery-phase studies, albeit at higher cost and technical complexity. Enzyme cycling assays provide robust, accessible, and cost-effective solutions for high-throughput screening and well-characterized biological systems. Future directions point toward hybrid approaches, the development of more stable isotopic internal standards, and in vivo sensing technologies. For the scientific community, a nuanced understanding of both methods' strengths and limitations, as outlined in this comparison, is essential for generating reliable, reproducible data that advances our understanding of redox biology and accelerates therapeutic innovation in metabolic diseases, cancer, and aging.