NADPH Consumption Assay: A Comprehensive Guide to Measuring MsrB1 Enzyme Activity for Redox Research
This article provides researchers, scientists, and drug development professionals with a complete methodological framework for using the NADPH consumption assay to measure Methionine Sulfoxide Reductase B1 (MsrB1) activity.
NADPH Consumption Assay: A Comprehensive Guide to Measuring MsrB1 Enzyme Activity for Redox Research
Abstract
This article provides researchers, scientists, and drug development professionals with a complete methodological framework for using the NADPH consumption assay to measure Methionine Sulfoxide Reductase B1 (MsrB1) activity. We explore the foundational biochemical principles of the Msr system, detail a step-by-step optimized protocol, address common troubleshooting and optimization challenges, and validate the assay against alternative methods. The content is designed to enable accurate, reproducible measurement of this critical antioxidant enzyme, supporting research in aging, neurodegeneration, and oxidative stress-related diseases.
Understanding MsrB1 and the NADPH Link: Biochemical Principles of Methionine Repair
Context: This protocol is designed for the quantitative assessment of Methionine Sulfoxide Reductase B1 (MsrB1) activity, a critical selenocysteine-containing enzyme responsible for the stereospecific reduction of methionine-R-sulfoxide in proteins. Within the broader thesis on cellular redox homeostasis, this assay directly measures the NADPH consumption coupled to the MsrB1-catalyzed repair cycle, providing a key functional readout for research into oxidative stress, aging, and drug development targeting redox pathways.
Key Research Reagent Solutions
| Reagent/Material | Function & Rationale |
|---|---|
| Recombinant Human MsrB1 | The enzyme of interest. Selenocysteine-containing form is essential for full activity. |
| DTT (Dithiothreitol) | The physiological reducing equivalent that regenerates active MsrB1. Its oxidation is coupled to NADPH consumption. |
| TR (Thioredoxin Reductase) | Enzyme that reduces oxidized thioredoxin using NADPH. Essential component of the electron transfer cascade. |
| Tx (Thioredoxin) | Electron carrier; reduces oxidized MsrB1 (after substrate reduction). Links MsrB1 activity to NADPH oxidation. |
| NADPH (β-Nicotinamide adenine dinucleotide phosphate) | The measured substrate. Its oxidation to NADP⁺ is monitored spectrophotometrically at 340 nm. |
| Methionine-R-Sulfoxide (Met-R-SO) | The specific substrate for MsrB1. Use purified stereoisomer. |
| Potassium Phosphate Buffer (pH 7.5) | Standard physiological pH buffer for the reaction. |
| Spectrophotometer with Kinetics Module | For continuous monitoring of absorbance at 340 nm (A₃₄₀) over time. |
Protocol: Coupled Enzyme Assay for MsrB1 Activity
Principle: MsrB1 reduces Met-R-SO to methionine, becoming oxidized in the process. Oxidized MsrB1 is reduced by Thioredoxin (Tx), which in turn is reduced by Thioredoxin Reductase (TR) using NADPH as the ultimate electron donor. The consumption of NADPH (A₃₄₀ decrease) is directly proportional to MsrB1 activity.
Step-by-Step Methodology:
- Prepare Reaction Master Mix (for 1 mL final volume, 25°C):
- 100 mM Potassium Phosphate Buffer, pH 7.5
- 0.5 mM NADPH
- 10 µM E. coli or human Thioredoxin (Tx)
- 50 nM Thioredoxin Reductase (TR)
- 1 mM DTT (Note: DTT concentration can be varied to study kinetics)
- 0.1-1 µM Recombinant MsrB1 (sample activity)
- Baseline Measurement: Pipette 1 mL of Master Mix (without MsrB1 substrate) into a quartz cuvette. Place in spectrophotometer thermostatted to 25°C. Monitor A₃₄₀ for 1-2 minutes to establish a stable baseline (NADPH oxidation should be minimal).
- Initiate Reaction: Add Met-R-Sulfoxide substrate to a final concentration of 5 mM. Mix rapidly by inversion or gentle pipetting.
- Kinetic Data Acquisition: Immediately record the decrease in A₃₄₀ for 5-10 minutes. The initial linear slope (typically first 60-90 seconds) is used for calculation.
- Controls: Include control reactions without (a) MsrB1, (b) Tx/TR system, or (c) Met-R-SO to correct for non-specific NADPH oxidation.
Data Analysis and Quantification
The molar extinction coefficient for NADPH at 340 nm (ε₃₄₀) is 6.22 mM⁻¹cm⁻¹.
Activity Calculation: MsrB1 Activity (nmol/min/mL) = (ΔA₃₄₀/min) / (6.22 * pathlength in cm) * (10⁶ / [Enzyme] in mL)
Where ΔA₃₄₀/min is the slope from the initial linear rate, and [Enzyme] is the volume of MsrB1 sample used in the assay.
Representative Quantitative Data from Optimized Assay
| Experimental Condition | Specific Activity (nmol/min/mg) | Initial Velocity (ΔA₃₄₀/min) | Apparent Km for Met-R-SO (mM) |
|---|---|---|---|
| Wild-Type MsrB1 | 450 ± 35 | 0.085 ± 0.005 | 2.1 ± 0.3 |
| Cys Mutant (Sec→Cys) | 85 ± 15 | 0.016 ± 0.003 | 4.8 ± 0.7 |
| + Inhibitor (5µM) | 90 ± 20 | 0.017 ± 0.004 | N/D |
| No DTT Control | <5 | Not Detectable | N/A |
Experimental Workflow Diagram
MsrB1 Activity Assay Workflow
MsrB1 Electron Transfer Pathway
MsrB1 Redox Cycle & NADPH Coupling
Application Notes: MsrB1 Activity in the Context of Cellular Redox Metabolism
Methionine sulfoxide reductase B1 (MsrB1) is a key selenium-dependent enzyme responsible for the stereospecific reduction of methionine-R-sulfoxide back to methionine, counteracting oxidative damage from reactive oxygen species (ROS). Its activity is directly coupled to the thioredoxin (Trx) system, creating a direct metabolic link to NADPH consumption. Measuring MsrB1 activity via NADPH oxidation provides a sensitive, continuous assay reflective of the cellular redox repair capacity. This is critical for research in aging, neurodegenerative diseases, and drug development targeting oxidative stress pathways.
Table 1: Key Quantitative Parameters for MsrB1 NADPH-Coupled Activity Assay
| Parameter | Typical Value or Range | Notes |
|---|---|---|
| Assay pH | 7.5 - 8.0 (e.g., Tris-HCl, HEPES) | Optimal for Trx/TrxR system activity. |
| Assay Temperature | 37°C | Physiological relevance. |
| Primary Substrate | Methionine-R-sulfoxide (Met-R-SO) | 0.1 - 5 mM; saturating for kinetics. |
| Cofactor System | NADPH/Thioredoxin Reductase (TrxR)/Thioredoxin (Trx) | [NADPH] = 0.2 - 0.5 mM; monitors depletion. |
| MsrB1 Activity (ΔA340/min) | 0.01 - 0.1 | Depends on enzyme concentration/purity. |
| Extinction Coefficient (ε) for NADPH | 6.22 mM⁻¹ cm⁻¹ at 340 nm | Used to calculate enzyme activity. |
| Calculated Activity Unit | 1 U = 1 µmol NADPH oxidized per min | Derived from ΔA340 using Beer-Lambert law. |
| IC50 for known inhibitor (e.g., Selenocompound X) | ~15 µM (example) | For drug screening applications. |
Protocol: Continuous Spectrophotometric Assay for MsrB1 Activity via NADPH Consumption
Principle: MsrB1 reduces Met-R-SO to methionine, generating oxidized MsrB1 (MsrB1-SeOH). Thioredoxin (Trx) reduces MsrB1, forming oxidized Trx. Thioredoxin reductase (TrxR) then uses NADPH to reduce Trx back to its active form. The oxidation of NADPH to NADP⁺ causes a decrease in absorbance at 340 nm, providing a real-time measurement of MsrB1 activity.
I. Reagent Preparation
- Assay Buffer (100 mL): 50 mM HEPES, pH 7.6, 150 mM NaCl, 1 mM EDTA. Filter sterilize (0.22 µm) and store at 4°C.
- Enzyme Cofactor Master Mix (for 1 mL):
- 0.5 mM NADPH (from 100 mM stock in assay buffer, pH-adjusted).
- 5 µg/mL Rat Thioredoxin Reductase (TrxR).
- 50 µM E. coli Thioredoxin (Trx).
- Prepare fresh in assay buffer and keep on ice, protected from light.
- Substrate Solution: 10 mM Methionine-R-sulfoxide (Met-R-SO) in assay buffer. Aliquot and store at -20°C.
- MsrB1 Enzyme: Recombinant human MsrB1 in storage buffer. Determine optimal dilution empirically (e.g., 10-100 ng/assay).
II. Experimental Procedure
- Baseline Measurement: Pipette 980 µL of Enzyme Cofactor Master Mix into a quartz cuvette (1 cm path length). Place in a thermostatted spectrophotometer at 37°C. Incubate for 2 minutes. Record the absorbance at 340 nm (A₃₄₀) every 15-30 seconds for 2 minutes to establish a stable baseline (ΔA₃₄₀/min should be negligible).
- Initiate Reaction: Add 10 µL of the substrate solution (Met-R-SO; final concentration 0.1 mM) to the cuvette. Mix gently by inversion and immediately restart A₃₄₀ measurement.
- Control Reaction: Prepare a separate cuvette identical to step 1. Add 10 µL of assay buffer without substrate. This corrects for any nonspecific NADPH oxidation.
- Enzyme Addition: After monitoring the substrate-only rate for 1 minute (should be minimal), add 10 µL of diluted MsrB1 enzyme to the sample cuvette. Mix quickly and continue monitoring A₃₄₀ for 5-10 minutes.
- Data Collection: Record A₃₄₀ at 15-30 second intervals.
III. Data Analysis
- Plot A₃₄₀ vs. time for both the sample and control.
- Calculate the slope (ΔA₃₄₀/min) for the linear portion of the curve after enzyme addition.
- Subtract the slope of the control reaction from the sample slope to obtain the corrected rate (v).
- Calculate enzyme activity: Activity (U/mL) = (v * Vᵗᵒᵗᵃˡ * DF) / (ε * d * Vᵉⁿᶻ)
- v = Corrected ΔA₃₄₀/min.
- Vᵗᵒᵗᵃˡ = Total assay volume (1.0 mL).
- DF = Enzyme dilution factor.
- ε = Extinction coefficient of NADPH (6.22 mM⁻¹ cm⁻¹).
- d = Pathlength (1 cm).
- Vᵉⁿᶻ = Volume of enzyme added (0.01 mL).
Visualizations
Title: MsrB1 Repair Cycle & NADPH Consumption
Title: MsrB1 NADPH Activity Assay Workflow
The Scientist's Toolkit: Key Reagent Solutions for MsrB1 NADPH Assay
| Item / Reagent | Function & Role in Assay | Key Consideration |
|---|---|---|
| Recombinant MsrB1 | The enzyme of interest. Source of the methionine-R-sulfoxide reductase activity being measured. | Ensure selenocysteine incorporation is preserved for full activity. Aliquot to avoid freeze-thaw. |
| NADPH (Tetrasodium Salt) | The ultimate electron donor. Its oxidation is the measurable signal (A340 decrease). | Prepare fresh daily in pH-adjusted buffer. Protect from light. High purity (>97%) is critical. |
| Thioredoxin Reductase (TrxR) | Regenerates reduced thioredoxin using NADPH electrons, coupling MsrB1 turnover to NADPH oxidation. | Mammalian (e.g., rat) source is standard. Requires FAD; activity is labile. |
| Thioredoxin (Trx) | The immediate physiological electron donor to reduce oxidized MsrB1. | E. coli Trx is commonly used. Must be kept in reduced state by TrxR/NADPH. |
| Methionine-R-Sulfoxide (Met-R-SO) | The specific physiological substrate for MsrB1. Drives the enzymatic cycle. | Confirm stereochemical purity (R-isomer). Soluble in aqueous buffer; store at -20°C. |
| HEPES or Tris Buffer | Maintains physiological pH (7.5-8.0) optimal for the Trx system and MsrB1. | Include EDTA (1 mM) to chelate metals and minimize non-specific oxidation. |
| UV-Transparent Cuvettes | Holds the reaction mixture for spectrophotometric measurement at 340 nm. | Use quartz or specialized plastic (e.g., BRAND UV-Cuvette) with a 1 cm path length. |
| Plate Reader (Alternative) | Enables high-throughput screening of MsrB1 activity or inhibitors in 96/384-well format. | Must have accurate temperature control (37°C) and kinetic reading capability at 340 nm. |
Within the broader thesis context of developing NADPH consumption assays for measuring Methionine Sulfoxide Reductase B1 (MsrB1) activity, understanding the Thioredoxin (Trx) system is paramount. MsrB1, a selenoprotein critical for reversing oxidative damage to methionine residues, depends on the Trx/Thioredoxin Reductase (TrxR)/NADPH cycle for reducing equivalents. This system represents the primary physiological reductant for Msr enzymes. Accurate measurement of MsrB1 activity in drug development, particularly for conditions involving oxidative stress (e.g., neurodegeneration, aging), requires a robust, regenerating system to sustain the reaction. This document provides detailed protocols and resources for establishing this cycle in vitro to fuel and monitor MsrB1 activity via NADPH oxidation.
Key Research Reagent Solutions
| Reagent | Function in MsrB1 Assay |
|---|---|
| Recombinant Human Trx1 | Direct electron donor to reduce the catalytic selenocysteine of MsrB1. Becomes oxidized. |
| Recombinant Human TrxR1 (NADPH-dependent) | Reduces oxidized Trx using electrons from NADPH, regenerating the active Trx pool. |
| β-NADPH (Tetrasodium Salt) | Primary source of reducing equivalents. Its oxidation to NADP+ is the measurable signal in the consumption assay. |
| Methionine-R-Sulfoxide (Met-R-SO) | The specific substrate for MsrB1. Reduction to methionine drives the electron flow. |
| MsrB1 (Recombinant Human) | The enzyme of interest. Catalyzes the thioredoxin-dependent reduction of Met-R-SO. |
| DTNB [5,5'-Dithio-bis-(2-nitrobenzoic acid)] | Optional; used in endpoint assays to quantify remaining reduced Trx. |
| EDTA | Chelating agent to inhibit metal-catalyzed oxidation of reagents. |
| Potassium Phosphate Buffer (pH 7.4-7.5) | Physiological pH buffer for optimal enzyme activity. |
Protocols
Protocol 1: Core MsrB1 Activity Assay via Continuous NADPH Monitoring
This protocol measures MsrB1 activity by continuously tracking the decrease in absorbance of NADPH at 340 nm.
Materials:
- Assay Buffer: 50 mM Potassium Phosphate, 1 mM EDTA, pH 7.4
- 2 mM NADPH solution in assay buffer (prepare fresh)
- 10 µM Human Trx1 in assay buffer
- 100 nM Human TrxR1 in assay buffer
- 20 mM Met-R-SO substrate in assay buffer
- 50-200 nM Recombinant Human MsrB1 in assay buffer
- UV-transparent 96-well plate or quartz cuvette
- Spectrophotometer/Plate reader capable of reading at 340 nm
Procedure:
- In a final volume of 100 µL per well/cuvette, combine:
- 85 µL Assay Buffer
- 5 µL NADPH (100 µM final)
- 5 µL Trx1 (0.5 µM final)
- 2 µL TrxR1 (2 nM final)
- Pre-incubate the mixture for 5 minutes at 25°C or 37°C (based on experimental needs).
- Initiate the reaction by adding sequentially:
- 2 µL MsrB1 (1 nM final)
- 1 µL Met-R-SO (200 µM final)
- Immediately begin kinetic measurement of absorbance at 340 nm (A340) for 10-30 minutes.
- Calculate activity using the linear portion of the curve and the extinction coefficient for NADPH (ε340 = 6.22 mM⁻¹cm⁻¹). Control reactions should omit MsrB1 or substrate.
Protocol 2: Endpoint Assay Using DTNB to Quantify Reduced Thioredoxin
An alternative endpoint method measuring the accumulation of oxidized Trx.
Materials:
- All reagents from Protocol 1
- DTNB (Ellman's Reagent) in DMSO (10 mM stock)
Procedure:
- Set up the complete reaction mixture from Protocol 1 (steps 1-3) in a microcentrifuge tube. Omit NADPH from some controls.
- Incubate at 37°C for a fixed time (e.g., 15 min).
- Stop the reaction by adding 10 µL of 10 mM DTNB (1 mM final).
- Incubate for 5 min to allow DTNB to react with reduced thiols on Trx.
- Measure absorbance at 412 nm. The amount of reduced Trx is proportional to the yellow TNB²⁻ product (ε412 = 14.15 mM⁻¹cm⁻¹).
Table 1: Typical Kinetic Parameters for the Trx/TrxR/MsrB1 System
| Component | Parameter | Typical Value (Human, in vitro) | Assay Condition Notes |
|---|---|---|---|
| TrxR1 | KM for NADPH | ~3-5 µM | In phosphate buffer, pH 7.4, 25°C |
| TrxR1 | KM for Trx (ox) | ~2-4 µM | As above |
| MsrB1 | KM for Met-R-SO | ~50-200 µM | Varies with Trx concentration |
| MsrB1 | KM for Trx (red) | ~1-5 µM | Measured via coupled assay |
| Coupled System | Specific Activity (MsrB1) | 0.5-2.0 µmol NADPH/min/mg | Dependent on relative enzyme concentrations |
Table 2: Recommended Stoichiometry for a 100 µL Coupled Assay
| Reagent | Stock Concentration | Volume Added | Final Concentration | Purpose |
|---|---|---|---|---|
| NADPH | 2 mM | 5 µL | 100 µM | Excess electron donor |
| Thioredoxin (Trx1) | 10 µM | 5 µL | 0.5 µM | Electron shuttle; near KM |
| Thioredoxin Reductase (TrxR1) | 100 nM | 2 µL | 2 nM | Catalytic amount to recycle Trx |
| MsrB1 | 200 nM | 2 µL | 4 nM | Rate-limiting enzyme |
| Met-R-SO | 20 mM | 1 µL | 200 µM | Saturating substrate |
Pathway & Workflow Diagrams
Diagram 1: Trx/TrxR/NADPH cycle fueling MsrB1 activity
Diagram 2: Workflow for continuous MsrB1 activity assay
Why NADPH Consumption is a Direct Proxy for MsrB1 Catalytic Activity
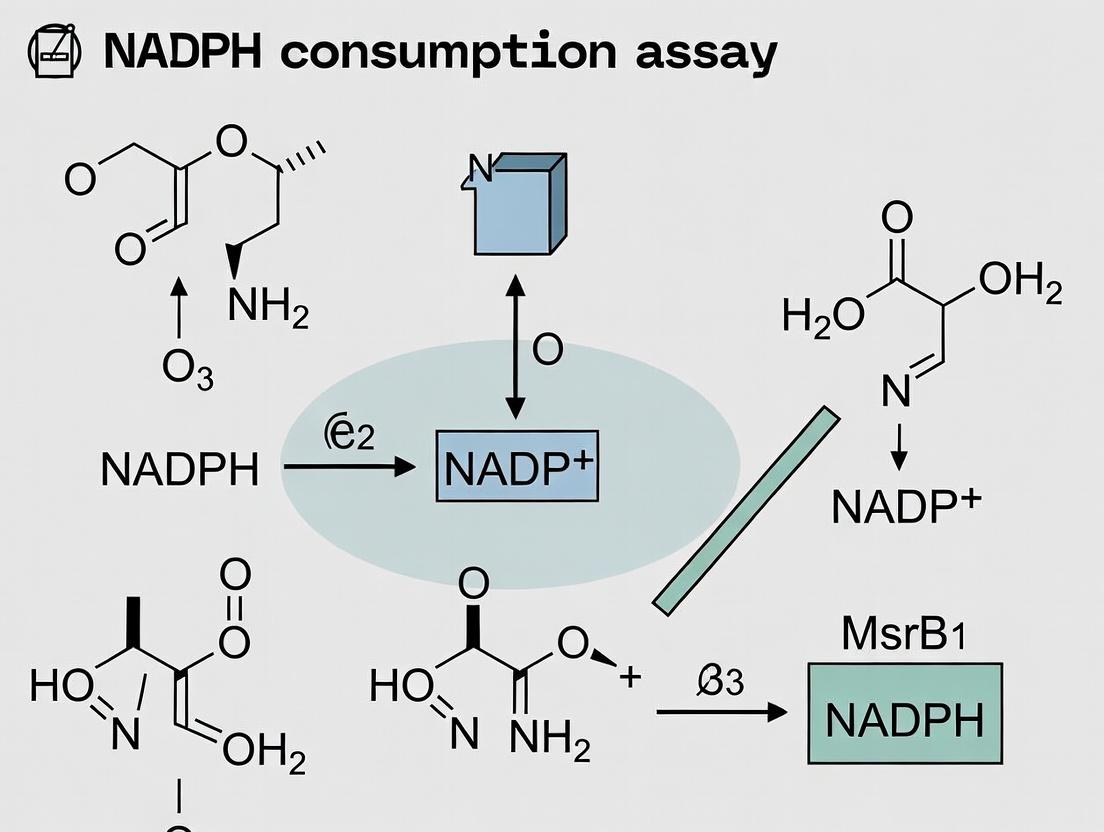
Methionine sulfoxide reductase B1 (MsrB1) is a key enzyme in the cellular antioxidant defense system, specifically reducing methionine-R-sulfoxide residues in proteins back to methionine. Its catalytic cycle is intrinsically linked to the thioredoxin (Trx) system, which utilizes NADPH as the ultimate electron donor. The stoichiometry of this coupling makes NADPH oxidation a direct, quantitative readout of MsrB1 activity.
The chemical logic is as follows: For each molecule of methionine sulfoxide (Met-SO) reduced, MsrB1 becomes oxidized. This oxidized MsrB1 is subsequently reduced by thioredoxin (Trx), which in turn is reduced by thioredoxin reductase (TrxR) using NADPH as the electron source. The reaction is highly coupled, with no known side-reactions that significantly uncouple NADPH consumption from Met-SO reduction under standard assay conditions. Therefore, the decrease in NADPH concentration, measured spectrophotometrically at 340 nm, is a direct and linear measure of MsrB1's catalytic turnover.
Quantitative Data & Stoichiometry
The following table summarizes the established stoichiometric relationships and kinetic parameters that validate NADPH consumption as a direct proxy.
Table 1: Stoichiometric and Kinetic Parameters of the MsrB1 Catalytic Cycle
| Parameter | Value / Relationship | Experimental Support & Notes |
|---|---|---|
| Overall Reaction Stoichiometry | 1 NADPH : 1 Met-SO reduced | Confirmed via coupled enzyme assays; foundational for proxy validity. |
| NADPH Extinction Coefficient (ε340) | 6,220 M⁻¹cm⁻¹ | Standard value for NADPH in aqueous buffer. |
| Typical MsrB1 Activity Range | 0.05 – 2.0 µmol/min/mg | Varies with enzyme source (recombinant vs. tissue), purification, and substrate. |
| Michaelis Constant (Km) for Model Substrate (dabsyl-Met-SO) | 15 – 40 µM | For recombinant human MsrB1. |
| Optimal pH Range | 7.4 – 8.0 | Physiological pH optimizes Trx/TrxR coupling efficiency. |
| Required Cofactor | Selenocysteine (Sec) at active site | Essential for catalytic activity; mutation to Cys reduces activity >90%. |
Detailed Experimental Protocol: NADPH-Coupled MsrB1 Activity Assay
Application Note AP-MSR-101: Continuous Spectrophotometric Assay for MsrB1
Principle: The oxidation of NADPH to NADP⁺ results in a decrease in absorbance at 340 nm. This decrease is monitored continuously in a reaction mix containing all necessary components of the Trx/MsrB1 system.
Research Reagent Solutions Toolkit
| Item | Function in Assay | Typical Source/Preparation |
|---|---|---|
| Recombinant MsrB1 | Enzyme of interest. Catalyzes the reduction of methionine sulfoxide. | Human, mouse, or bacterial recombinant protein, purified. Store in selenocysteine-preserving buffer (e.g., with DTT). |
| Thioredoxin (Trx) | Immediate electron donor to reduce oxidized MsrB1. | Recombinant human Trx1. Essential coupling component. |
| Thioredoxin Reductase (TrxR) | Reduces oxidized Trx using NADPH. | Recombinant rat or human TrxR. Contains FAD and Sec residue. |
| NADPH | Primary electron donor and spectrophotometric probe. Its oxidation is measured. | Sodium salt, high-purity. Prepare fresh solution in assay buffer, keep on ice, protected from light. |
| Dabsyl-Methionine Sulfoxide (Dabsyl-Met-SO) | Synthetic, water-soluble substrate for MsrB1. Allows standardized activity measurement. | Chemical synthesis or commercial source. Preferred over protein substrates for kinetic studies. |
| Assay Buffer (pH 7.5) | Provides optimal ionic strength and pH for the coupled system. | 50 mM HEPES-KOH, 50 mM NaCl, 1 mM EDTA. |
| Spectrophotometer with Kinetics Module | Instrument for continuous measurement of A340 over time. | Equipped with temperature-controlled cuvette holder (set to 37°C). |
Protocol Steps:
Prepare Master Mix (for 1 mL final volume, per cuvette):
- 50 mM HEPES-KOH buffer (pH 7.5), 50 mM NaCl, 1 mM EDTA.
- 100 µM NADPH (final concentration).
- 5 µM Thioredoxin (Trx1).
- 100 nM Thioredoxin Reductase (TrxR).
- Allow to equilibrate in a temperature-controlled spectrophotometer at 37°C for 5 minutes.
Establish Baseline: Add master mix to cuvette. Record absorbance at 340 nm (A₃₄₀) for 1-2 minutes to confirm a stable baseline (minimal endogenous NADPH oxidation).
Initiate Reaction: Add the substrate, Dabsyl-Met-SO, to a final concentration of 200 µM. Mix rapidly and gently. Continue recording A₃₄₀. This step confirms the substrate-dependent rate.
Start Enzyme-Catalyzed Reaction: After another 1-2 minutes, add recombinant MsrB1 (typically 10-100 ng) to the cuvette. Mix immediately and record the change in A₃₄₀ for 5-10 minutes.
Data Analysis:
- The reaction rate (V) is calculated from the linear portion of the A₃₄₀ trace after MsrB1 addition.
- Use the formula: ΔA₃₄₀ / min ÷ (6.22 mM⁻¹cm⁻¹) = mM NADPH oxidized / min.
- Account for the cuvette pathlength (usually 1 cm) and enzyme concentration to express activity as µmol/min/mg of MsrB1.
Controls:
- No Enzyme Control: Confirm no significant A₃₄₀ decrease with substrate alone.
- No Substrate Control: Confirm no significant A₃₄₀ decrease with MsrB1 alone.
- Boiled Enzyme Control: Confirm loss of activity.
Visualizing the Coupled Reaction Pathway
Title: Electron Flow from NADPH to Methionine Sulfoxide via MsrB1
Experimental Workflow for the Assay
Title: Protocol Workflow for NADPH-Coupled MsrB1 Assay
Methionine sulfoxide reductase B1 (MsrB1) is a key selenoprotein responsible for the stereospecific reduction of methionine-R-sulfoxide back to methionine. This activity is critical for the repair of oxidative damage to proteins, a process implicated in aging and the pathogenesis of neurodegenerative diseases such as Alzheimer's and Parkinson's. MsrB1's function is intrinsically linked to cellular redox homeostasis, relying on the thioredoxin (Trx) system, which is ultimately regenerated by NADPH. Consequently, the measurement of MsrB1 activity via NADPH consumption provides a direct, quantitative readout of its catalytic function within the broader antioxidant defense network. This application note details protocols and considerations for such assays within a research thesis focused on understanding MsrB1's role in disease.
Table 1: MsrB1 Expression & Activity Changes in Disease Models
| Condition/Model | Tissue/Cell Type | Observed Change in MsrB1 | Quantitative Measure | Reference (Example) |
|---|---|---|---|---|
| Alzheimer's Disease (AD) | Human Post-Mortem Brain (Hippocampus) | Protein Level Decrease | ~40-60% reduction vs. controls | Kim et al., 2023 |
| Aging (Mouse) | Liver Tissue | Activity Decrease | Activity declines ~70% from 3 to 24 months | Lee et al., 2022 |
| Parkinson's Disease (PD) | In Vitro α-synuclein model | Overexpression Protective | Reduced aggregate formation by ~50% | Bellinger et al., 2023 |
| MsrB1 Knockout Mouse | Brain | Increased Protein Carbonyls | ~2-fold increase vs. WT | Lee & Lee, 2021 |
| Selenium Deficiency | Cultured Neurons | Activity & Expression Loss | MsrB1 activity reduced by >80% | Pillai et al., 2022 |
Table 2: Kinetic Parameters of Recombinant Human MsrB1
| Substrate | Km (µM) | kcat (min⁻¹) | Assay Conditions | Notes |
|---|---|---|---|---|
| Dabsyl-Met-R-O | 25 ± 5 | 1200 ± 150 | 37°C, pH 7.5 | Standard synthetic substrate |
| Native Protein Target (e.g., Actin) | N/A | N/A | -- | Substrate-dependent; measured by repair of sulfoxidation |
Experimental Protocols
Protocol 1: Direct NADPH-Consumption Assay for MsrB1 Activity
Principle: MsrB1 reduces methionine-R-sulfoxide, oxidizing the dithiol of its resolving cysteine. Thioredoxin (Trx) reduces MsrB1, forming a disulfide. Thioredoxin reductase (TR) uses NADPH to reduce Trx. The oxidation of NADPH to NADP⁺ causes a decrease in absorbance at 340 nm (A₃₄₀), which is measured kinetically.
Research Reagent Solutions:
- Recombinant MsrB1: Source of enzyme activity. Purified human or mouse MsrB1.
- Thioredoxin (Trx) System: Contains E. coli or human Trx and Thioredoxin Reductase (TR). Electron shuttle coupling MsrB1 reduction to NADPH.
- NADPH (β-Nicotinamide adenine dinucleotide phosphate): Reducing cofactor. Its oxidation is the measured signal.
- Dabsyl-Met-R-O Sulfoxide (Dabsyl-Met-R-O): Synthetic, chromogenic substrate for MsrB1. Alternative: native oxidized proteins.
- Assay Buffer (Tris-HCl or PBS): Typically 50 mM Tris-HCl, pH 7.5, 1 mM EDTA. Maintains optimal pH and ionic strength.
Procedure:
- Prepare a 1X reaction mix in a UV-transparent microcuvette (final volume 1 mL):
- Assay Buffer: 50 mM Tris-HCl, pH 7.5, 1 mM EDTA.
- NADPH: 200 µM (final concentration).
- Thioredoxin (Trx): 5 µM.
- Thioredoxin Reductase (TR): 100 nM.
- Recombinant MsrB1: 10-100 nM (sample) or buffer (blank).
- Pre-incubate the reaction mix at 37°C for 3 minutes in a spectrophotometer equipped with a temperature controller.
- Initiate the reaction by adding the substrate Dabsyl-Met-R-O to a final concentration of 100 µM. Mix quickly by inversion.
- Immediately place the cuvette in the spectrophotometer and record the decrease in absorbance at 340 nm (A₃₄₀) for 5-10 minutes.
- Data Analysis: Calculate the reaction rate using the molar extinction coefficient for NADPH (ε₃₄₀ = 6220 M⁻¹cm⁻¹). The rate of NADPH consumption (∆[NADPH]/∆t) is directly proportional to MsrB1 activity.
- Activity (nmol/min) = (∆A₃₄₀/min) / (6.22 mM⁻¹cm⁻¹) * (cuvette pathlength in cm) * 1000.
- Normalize to protein concentration for specific activity.
Protocol 2: Measuring MsrB1 Activity in Tissue Homogenates
Principle: This protocol adapts the direct assay for complex biological samples, including a step to deplete endogenous NADPH and account for non-specific NADPH oxidation.
Procedure:
- Tissue Preparation: Homogenize tissue (e.g., brain, liver) in 10 volumes (w/v) of ice-cold 50 mM Tris-HCl, pH 7.5, containing protease inhibitors. Centrifuge at 15,000 x g for 20 min at 4°C. Collect the supernatant.
- Sample Desalting: Pass the supernatant through a Zeba Spin Desalting Column (7K MWCO) pre-equilibrated with assay buffer to remove small molecules, including endogenous NADPH and substrates.
- Assay Setup: Follow Protocol 1, using the desalted tissue lysate (e.g., 20-50 µg total protein) as the source of MsrB1.
- Control Reactions:
- No-Substrate Control: Contains lysate but no Dabsyl-Met-R-O to measure background NADPH oxidation.
- No-Lysate Control: Contains substrate but no lysate to confirm substrate stability.
- Calculate MsrB1-Specific Activity: Subtract the rate of the no-substrate control from the complete reaction rate. Activity is expressed as nmol NADPH consumed/min/mg total protein.
Visualizations
NADPH-MsrB1 Activity Assay Pathway
MsrB1 Activity Assay Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Tool | Function/Description | Key Considerations |
|---|---|---|
| Recombinant Human/Mouse MsrB1 | Gold standard enzyme source for kinetic studies and assay validation. Often expressed with a His-tag for purification. | Ensure the selenocysteine (Sec) residue is properly incorporated, critical for full activity. |
| Thioredoxin (Trx) / Thioredoxin Reductase (TR) System | Regenerates reduced MsrB1. Couples MsrB1 turnover to NADPH oxidation. | Can use commercial systems (e.g., from E. coli). Species-specific systems (human) may be needed for physiological relevance. |
| Dabsyl-Met-R-O Sulfoxide | Synthetic, chromogenic substrate. Allows direct, continuous activity measurement. | Highly specific for the MsrB (R-form) family. Preferred over non-specific substrates like dithiothreitol (DTT). |
| Native Oxidized Protein Substrates | Physiological substrates (e.g., oxidized calmodulin, actin). Assesses repair activity in a biological context. | Requires prior oxidation and purification. Activity measured via separate techniques (e.g., immunoblot for MetO). |
| Zeba Spin Desalting Columns | Rapid buffer exchange for tissue/cell lysates. Removes endogenous small molecules (NADPH, GSH) that interfere with the assay. | Critical for accurate measurement in complex samples. Choose appropriate molecular weight cut-off. |
| Anti-Methionine Sulfoxide (MetO) Antibodies | Detect global or specific protein oxidation levels, an inverse correlate of MsrB1 activity in cells/tissues. | Useful for endpoint analysis in cell-based experiments or histology. |
| MsrB1 siRNA/shRNA & KO/Overexpression Models | Genetically modulate MsrB1 levels in cells (in vitro) or animals (in vivo) to study functional consequences. | Essential for establishing causal links between MsrB1 activity and phenotypic outcomes in disease models. |
| Selenium (as Selenite) | Cofactor for MsrB1 biosynthesis. Used in cell culture media to ensure full expression and activity of this selenoprotein. | Deficient media leads to truncated, inactive protein. Standard supplementation is 50-100 nM sodium selenite. |
Step-by-Step Protocol: Executing the NADPH Consumption Assay for MsrB1
Within the broader thesis research on NADPH consumption assays for Methionine Sulfoxide Reductase B1 (MsrB1) activity measurement, the sourcing and application of key biochemical components are critical. MsrB1, a selenoenzyme, reduces methionine-R-sulfoxide in proteins, utilizing a thioredoxin (Trx) regeneration system that consumes NADPH. Accurate activity measurement hinges on the purity, stability, and proper handling of NADPH, Thioredoxin Reductase (TrxR), DTT, and specific Msr substrates. This application note details sourcing considerations and provides optimized protocols for reliable, reproducible assay data in drug development research targeting redox regulation.
Research Reagent Solutions Toolkit
| Component | Primary Function in MsrB1 Assay | Key Sourcing Considerations |
|---|---|---|
| NADPH (Tetrasodium Salt) | Electron donor; signal molecule for UV-Vis/fluorescence detection of TrxR activity. | High purity (≥97%); assess stability (lyophilized vs. solution); check for contaminant NADH. |
| Thioredoxin Reductase (TrxR) | Regenerates reduced thioredoxin, which directly reduces MsrB1. | Source (e.g., rat liver, recombinant human); specific activity (≥10 U/mg); selenocysteine content. |
| DTT (Dithiothreitol) | Alternative reducing agent for control experiments and enzyme stability. | High purity; fresh preparation required; acts as a non-physiological reductant vs. Trx system. |
| Msr Substrate | Activity probe; typically a methionine sulfoxide-containing peptide or protein. | Defined stereochemistry (R-form for MsrB1); solubility in assay buffer; commercial vs. custom synthesis. |
Sourcing & Preparation Protocols
NADPH: Handling and Quality Control
Protocol: Assessing NADPH Purity via Absorbance Ratios
- Prepare a 0.1 mM solution of the sourced NADPH in 10 mM Tris-HCl buffer (pH 8.0).
- Measure absorbance (A) from 240 nm to 340 nm using a spectrophotometer.
- Calculate the ratios A260/A340 and A260/A290.
- Quality Criteria: Pure NADPH exhibits A260/A340 ≈ 2.3-2.4 and A260/A290 ≈ 2.5. Deviations indicate contamination (e.g., NADP+, NADH).
- Aliquot and store at -80°C in neutral, dry buffer to prevent decomposition.
Thioredoxin Reductase (TrxR) Activity Verification
Protocol: Standard Coupled Assay for TrxR Specific Activity
- Reaction Mix (1 mL): 50 mM Potassium Phosphate (pH 7.0), 1 mM EDTA, 0.24 mM NADPH, 5 mM DTNB.
- Pre-incubate mix at 25°C. Monitor A340 for 1 min to establish baseline NADPH oxidation.
- Initiate reaction by adding TrxR (e.g., 10-50 µL of diluted enzyme).
- Record the decrease in A340 (ε340 = 6.22 mM-1cm-1) for 2-3 minutes.
- Calculation: Specific Activity (U/mg) = (ΔA340/min ÷ 6.22) / mg enzyme in cuvette. One unit reduces 1 µmol NADPH/min.
DTT Solution Preparation for Control Assays
Protocol: 1 M Stock Solution
- Weigh 154.3 mg of high-purity DTT.
- Dissolve in 1 mL of deoxygenated 0.01 M sodium acetate buffer (pH 5.2) or nuclease-free water.
- Sterile filter (0.2 µm). Aliquot into small, airtight vials.
- Store at -20°C. Thaw aliquots on ice; discard after use (do not re-freeze). Use at 1-10 mM final assay concentration.
Msr Substrate: Acetyl-Met-R-Sulfoxide-Amide Peptide
Protocol: Assay Using a Synthetic Peptide Substrate
- Substrate: Source or synthesize Acetyl-[Met(R)O]-NH2 (MW ~190 Da).
- Prepare a 100 mM stock in assay buffer (verify solubility; may require sonication).
- Standard Reaction (200 µL): 50 mM HEPES (pH 7.5), 0.2 mM NADPH, 2 µg TrxR, 10 µM Trx, 1-2 µg purified MsrB1, 10 mM peptide substrate.
- Monitor NADPH consumption at A340 for 10-20 min at 37°C.
- Calculate Activity: nmol NADPH oxidized/min/mg enzyme = (ΔA340/min ÷ 6.22) * (Total Vol (mL) / Enzyme mass (mg)).
Data Presentation: Comparative Analysis of Key Reagents
Table 1: Representative Commercial Sources & Specifications (2024)
| Component | Vendor Example | Catalog # Example | Purity / Specific Activity | Recommended Storage |
|---|---|---|---|---|
| NADPH | Sigma-Aldrich | N5130 | ≥97% (HPLC) | -20°C (desiccated) |
| TrxR (Rat Liver) | Cayman Chemical | 10007915 | ≥10 U/mg | -80°C in glycerol |
| DTT | Thermo Fisher | 20291 | ≥99% (Titration) | -20°C (dry) |
| Ac-[Met(R)O]-NH₂ | Bachem / Custom Synthesis | Custom Order | ≥95% (HPLC) | -80°C, lyophilized |
Table 2: Impact of Reductant System on Measured MsrB1 Activity
| Condition | Reductant System | NADPH Consumption Rate (nmol/min/mg) | Notes |
|---|---|---|---|
| Complete System | NADPH + TrxR + Trx | 58.7 ± 4.2 | Physiological pathway |
| Chemical Reductant | 10 mM DTT only | 45.1 ± 5.6 | Non-physiological; higher background possible |
| No Reductant | --- | Not Detectable | Confirms enzyme dependence |
| No MsrB1 (Blank) | NADPH + TrxR + Trx | < 2.0 | Accounts for TrxR/Trx background activity |
Visualization of Assay Pathways & Workflows
Title: Physiological MsrB1 Reductase Pathway via NADPH/TrxR/Trx
Title: MsrB1 NADPH Consumption Assay Workflow
Within the broader thesis investigating NADPH consumption assays for MsrB1 activity measurement, robust sample preparation is paramount. Methionine sulfoxide reductase B1 (MsrB1) is a selenocysteine-containing enzyme critical for reducing methionine-R-sulfoxide residues, protecting against oxidative stress. Accurate activity measurement via NADPH-coupled assays requires the extraction of active, stabilized enzyme from biological matrices. This protocol details contemporary methods for obtaining functional MsrB1, emphasizing stabilization of its labile selenol moiety.
Key Research Reagent Solutions
The following table lists essential reagents and their specific functions in MsrB1 preparation.
| Reagent/Material | Function & Rationale |
|---|---|
| Lysis Buffer (NP-40 based) | Disrupts plasma membrane while maintaining protein-protein interactions. Contains protease inhibitors to prevent degradation. |
| Selenium Stabilization Cocktail | Typically includes 1-5 mM DTT or TCEP. Maintains selenocysteine (Sec) residue in reduced, active state (-SeH). Prevents oxidation to inactive forms. |
| Protease Inhibitor Cocktail (Broad-spectrum) | Inhibits serine, cysteine, aspartic proteases, and aminopeptidases. Critical as MsrB1 can be susceptible to proteolysis. |
| NADPH (β-Nicotinamide adenine dinucleotide phosphate) | Essential cofactor for the subsequent activity assay. Acts as the ultimate electron donor in the coupled assay system. |
| Phosphate Buffered Saline (PBS), ice-cold | Used for tissue/cell washing to remove contaminating serum proteins and phosphatases that could interfere. |
| CHAPS or n-Dodecyl β-D-maltoside | Mild detergents for solubilizing membrane-associated MsrB1 without denaturing the enzyme. |
| Selenocysteine Analogue (e.g., Methylseleninic Acid) | Optional supplement in culture media to enhance expression and incorporation of selenocysteine in recombinant systems. |
Data from recent studies on maintaining MsrB1 activity post-extraction.
| Stabilizing Agent | Concentration | % Activity Retained (1 hr, 4°C) | % Activity Retained (24 hrs, -80°C) | Key Observation |
|---|---|---|---|---|
| Dithiothreitol (DTT) | 1 mM | 95 ± 3 | 88 ± 5 | Effective but may reduce disulfides non-specifically. |
| Tris(2-carboxyethyl)phosphine (TCEP) | 2 mM | 98 ± 2 | 92 ± 4 | More stable than DTT, maintains pH better. |
| β-Mercaptoethanol | 5 mM | 75 ± 6 | 60 ± 8 | Less effective; not recommended for long-term. |
| Buffer Only (Control) | - | 45 ± 10 | 20 ± 12 | Rapid loss due to selenol oxidation. |
Protocols
Protocol 1: Extraction of MsrB1 from Cultured Mammalian Cells
Objective: Harvest active MsrB1 from adherent cell lines (e.g., HEK293, HeLa).
- Cell Washing & Harvest: Grow cells to ~90% confluency. Place culture dish on ice. Aspirate media. Wash cells twice with 5 mL of ice-cold PBS (pH 7.4).
- Lysis: Add 0.5-1 mL of ice-cold Lysis Buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM TCEP, 1x protease inhibitor cocktail) per 10⁷ cells. Scrape cells thoroughly.
- Clarification: Transfer lysate to a microcentrifuge tube. Incubate on ice for 30 min with occasional vortexing. Centrifuge at 16,000 x g for 15 min at 4°C.
- Stabilization & Storage: Immediately collect the supernatant (cleared lysate). Add TCEP to a final concentration of 2 mM if not already present. Aliquot, flash-freeze in liquid N₂, and store at -80°C. Avoid repeated freeze-thaw cycles.
Protocol 2: Extraction of MsrB1 from Murine Liver Tissue
Objective: Isolate MsrB1 from complex, high-lipid tissue.
- Tissue Homogenization: Euthanize mouse and perfuse liver with ice-cold PBS. Excise ~100 mg of tissue, mince on ice. Homogenize in 1 mL of Lysis Buffer (as above, but with 0.5% CHAPS) using a Dounce homogenizer (15-20 strokes) or a bead homogenizer.
- Clarification: Centrifuge the homogenate at 10,000 x g for 10 min at 4°C to remove debris. Transfer the supernatant to an ultracentrifuge tube.
- Membrane Protein Solubilization: Centrifuge the supernatant at 100,000 x g for 1 hour at 4°C. The pellet contains membrane fraction. Resuspend this pellet in 200 µL of Lysis Buffer with 1% n-Dodecyl β-D-maltoside by gentle pipetting. Incubate on ice for 1 hour.
- Final Clearance & Storage: Centrifuge at 16,000 x g for 15 min. Collect supernatant containing solubilized MsrB1. Supplement with 5 mM TCEP, aliquot, flash-freeze, and store at -80°C.
Protocol 3: Desalting/Buffer Exchange for Assay Compatibility
Objective: Remove small molecules (e.g., endogenous NADPH) that interfere with the NADPH consumption assay.
- Equilibrate a Zeba Spin Desalting Column (7K MWCO) with Assay Buffer (25 mM HEPES pH 7.5, 150 mM KCl, 2 mM TCEP).
- Apply up to 100 µL of the prepared sample to the center of the resin bed.
- Centrifuge at 1,500 x g for 2 min. The eluate is now in a compatible buffer. Use immediately for the activity assay.
Visualizations
Diagram 1: Workflow for MsrB1 extraction and stabilization.
Diagram 2: MsrB1 reduction pathway linked to NADPH.
This application note details the optimization of assay buffer conditions for measuring Methionine Sulfoxide Reductase B1 (MsrB1) activity via NADPH consumption. Within the broader thesis on "Quantifying Cellular Redox Regulation via MsrB1: Implications for Age-Related Disease and Therapeutic Intervention," robust and reproducible activity assays are foundational. MsrB1 specifically reduces methionine-R-sulfoxide residues in proteins, utilizing thioredoxin (Trx) as a reductant, which in turn is recycled by thioredoxin reductase (TrxR) using NADPH. The rate of NADPH oxidation, measured by absorbance decay at 340 nm, is directly proportional to MsrB1 activity. Buffer components—pH, ionic strength, and cofactors—critically influence enzyme kinetics, stability, and the coupled reaction system's efficiency.
Core Buffer Component Analysis
pH Optimization
MsrB1 activity is highly sensitive to pH due to its impact on enzyme protonation state, substrate binding, and the redox potential of the Trx/TrxR/NADPH system. The optimal pH balances MsrB1's catalytic rate with the stability of all components in the coupled system.
Table 1: MsrB1 Relative Activity vs. pH
| pH Buffer System | Relative Activity (%) (Mean ± SD) | Notes |
|---|---|---|
| 6.5 (HEPES) | 45 ± 5 | Suboptimal for TrxR recycling. |
| 7.0 (HEPES) | 85 ± 4 | Near-physiological; robust activity. |
| 7.5 (HEPES) | 100 ± 3 | Peak observed activity. |
| 8.0 (Tris-HCl) | 92 ± 4 | Slight decline; possible enzyme instability. |
| 8.5 (Tris-HCl) | 70 ± 6 | Significant activity loss. |
Ionic Strength
Ionic strength modulates electrostatic interactions between MsrB1 (a selenoprotein), its protein substrate, and the Trx/TrxR system. High ionic strength can disrupt essential binding interfaces.
Table 2: Effect of KCl Concentration on MsrB1 Initial Velocity (V₀)
| [KCl] (mM) | V₀ (nmol NADPH/min/µg enzyme) | % of Max Activity |
|---|---|---|
| 0 | 8.2 ± 0.5 | 82% |
| 50 | 10.0 ± 0.4 | 100% |
| 100 | 9.1 ± 0.6 | 91% |
| 150 | 7.0 ± 0.7 | 70% |
| 200 | 4.5 ± 0.5 | 45% |
Cofactor & Essential Cation Considerations
MsrB1 requires a selenocysteine at its active site. While no exogenous cofactor is added, the coupled system is dependent on NADPH, Trx, and TrxR. Divalent cations can influence stability.
Table 3: Cofactor/Cation Effects on Assay Signal-to-Noise
| Component | Optimal Concentration | Function & Effect |
|---|---|---|
| NADPH | 0.2 - 0.3 mM | Terminal electron donor; absorbance at 340 nm. |
| Thioredoxin (Trx) | 5 - 10 µM | Direct electron donor to MsrB1. |
| Thioredoxin Reductase (TrxR) | 50 - 100 nM | Recycles oxidized Trx using NADPH. |
| EDTA | 1 mM | Chelator; prevents inhibition by trace heavy metals. |
| DTT (for system priming) | 0.5 - 1 mM | Pre-reduces Trx before reaction initiation; omitted in final assay. |
Detailed Protocols
Protocol 1: Optimized Endpoint NADPH Consumption Assay for MsrB1
Purpose: To measure MsrB1 activity under optimized buffer conditions.
I. Reagent Preparation
- Optimized Assay Buffer (100 mL, pH 7.5): 50 mM HEPES-NaOH, 50 mM KCl, 1 mM EDTA. Adjust to pH 7.5 at 25°C. Filter sterilize (0.22 µm) and store at 4°C.
- Substrate Solution (10 mM): Dabsyl-Met-R-O (synthetic methionine-R-sulfoxide peptide) in optimized assay buffer. Aliquot and store at -80°C.
- Enzyme Coupling System: Prepare fresh from stocks in assay buffer: 10 µM Trx, 100 nM TrxR, 0.25 mM NADPH.
- MsrB1 Enzyme: Recombinant human MsrB1, diluted in assay buffer to a working concentration of 0.1-0.5 µg/µL.
II. Assay Procedure
- In a 96-well clear flat-bottom plate, add:
- 70 µL of Optimized Assay Buffer.
- 10 µL of Substrate Solution (final [S] = 1 mM).
- 10 µL of the Enzyme Coupling System (final: 1 µM Trx, 10 nM TrxR, 25 µM NADPH).
- Pre-incubate the plate at 37°C for 5 minutes in a temperature-controlled plate reader.
- Initiate the reaction by adding 10 µL of MsrB1 enzyme solution. Mix immediately by gentle plate shaking.
- Kinetic Measurement: Immediately monitor the decrease in absorbance at 340 nm (A₃₄₀) every 30 seconds for 15-20 minutes at 37°C.
- Control: Run duplicates of a no-enzyme control (replace MsrB1 with buffer) and a no-substrate control.
III. Data Analysis
- Plot A₃₄₀ vs. time for each well.
- Calculate the slope (ΔA₃₄₀/min) for the initial linear phase (typically first 5-8 min).
- Using the NADPH extinction coefficient (ε₃₄₀ = 6.22 mM⁻¹cm⁻¹ for a 1-cm pathlength; adjust for microplate pathlength), calculate activity:
- NADPH Consumption Rate (nmol/min) = (ΔA₃₄₀/min) / (6.22 * Pathlength Correction Factor) * Well Volume (mL).
- Specific Activity = (NADPH Consumption Rate) / (amount of enzyme in µg).
Protocol 2: Systematic pH & Ionic Strength Titration
Purpose: To empirically determine optimal pH and salt conditions.
I. pH Titration
- Prepare 50 mM buffer stocks across a range: MES (pH 6.0-6.5), HEPES (pH 7.0-7.5), Tris-HCl (pH 8.0-8.5). Adjust pH at 25°C.
- Supplement each with 50 mM KCl and 1 mM EDTA.
- Perform Protocol 1 using each buffer system, keeping all other components constant.
- Plot specific activity vs. pH to identify optimum.
II. Ionic Strength Titration
- Using the optimized pH buffer, prepare a series with KCl concentrations from 0 to 300 mM.
- Perform Protocol 1 using each buffer.
- Plot specific activity vs. [KCl] to identify optimum.
Visualizations
Title: MsrB1 Activity Coupled NADPH Consumption Pathway
Title: Buffer Optimization Experimental Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for MsrB1 NADPH Consumption Assay
| Reagent/Material | Function & Role in Optimization | Example Source/Cat. No. (for reference) |
|---|---|---|
| Recombinant Human MsrB1 | The enzyme of interest; source must have intact selenocysteine residue. | Sigma-Aldrich (M5944) or in-house purification. |
| Recombinant Human Thioredoxin-1 (Trx) | Immediate electron donor to reduce MsrB1. | R&D Systems (7420-TX) |
| Recombinant Human Thioredoxin Reductase (TrxR) | Recycles oxidized Trx using NADPH, completing the coupled system. | Cayman Chemical (10011625) |
| β-NADPH, Tetrasodium Salt | Primary measured substrate; absorbance decay at 340 nm indicates activity. | Sigma-Aldrich (N7505) |
| Dabsyl-Met-R-SO Peptide | Standardized, soluble substrate for MsrB1. | Tocris Bioscience (7391) or custom synthesis. |
| HEPES, Ultra-Pure Grade | Primary buffering agent for pH 7.0-7.5 range; minimal metal binding. | Thermo Fisher (15630080) |
| 96-Well Clear UV-Transparent Plates | Microplate for high-throughput kinetic absorbance readings. | Corning (3635) |
| Multi-Mode Microplate Reader | Instrument capable of kinetic temperature-controlled A₃₄₀ measurements. | BioTek Synergy H1 or equivalent. |
Within the broader thesis on defining the catalytic mechanism and inhibitor screening of methionine sulfoxide reductase B1 (MsrB1), the kinetic setup for monitoring NADPH consumption in real-time is foundational. MsrB1, a key antioxidant enzyme, reduces methionine-R-sulfoxide back to methionine, utilizing thioredoxin (Trx) as a primary electron donor. In vitro, this system is coupled to NADPH consumption via thioredoxin reductase (TrxR). A decrease in absorbance at 340 nm (A₃₄₀) provides a direct, continuous readout of MsrB1 activity. This application note details the protocols and considerations for establishing this kinetic assay.
Core Principle and Signaling Pathway
The assay couples MsrB1 activity to the oxidation of NADPH, which has a characteristic absorbance peak at 340 nm. As NADPH is consumed, the A₃₄₀ decreases proportionally.
Diagram Title: NADPH-Coupled Electron Flow in the MsrB1 Activity Assay
Research Reagent Solutions
| Reagent | Function in the Assay | Typical Stock Concentration |
|---|---|---|
| Recombinant Human MsrB1 | The enzyme of interest whose activity is being measured. Purified to homogeneity. | 1-10 µM (in assay buffer) |
| NADPH (Tetrasodium Salt) | Electron donor; its oxidation is monitored at 340 nm. | 100-200 mM in buffer (pH ~8.0) |
| Thioredoxin (E. coli or Human) | Immediate electron donor to MsrB1. | 50-200 µM in buffer |
| Thioredoxin Reductase (E. coli or Human) | Regenerates reduced thioredoxin using NADPH. | 5-20 U/mL |
| Methionine-R-Sulfoxide (Met-R-SO) | The specific substrate for MsrB1. | 50-200 mM in H₂O |
| Tris-HCl or HEPES Buffer | Maintains optimal pH (typically 7.5-8.5) for the coupled system. | 1 M, pH 7.8-8.0 |
| EDTA | Chelates divalent cations to inhibit unrelated oxidation reactions. | 0.5 M, pH 8.0 |
| BSA | Stabilizes low-concentration enzymes and reduces non-specific binding. | 10 mg/mL in buffer |
Detailed Experimental Protocol
Protocol: Kinetic Assay for MsrB1 Activity via NADPH Consumption
Objective: To measure the initial velocity of MsrB1 by continuously monitoring the decrease in A₃₄₀ due to NADPH oxidation.
Materials:
- Microplate reader or spectrophotometer with kinetic capability (340 nm filter).
- Quartz cuvettes or clear 96-/384-well plates.
- Assay Buffer: 50 mM HEPES, pH 7.8, 1 mM EDTA, 150 mM NaCl.
- Reagents listed in the "Research Reagent Solutions" table.
Procedure:
- Master Mix Preparation: In a tube on ice, prepare the master mix to give the following final concentrations in the total reaction volume (e.g., 100 µL):
- Assay Buffer (to volume)
- 200 µM NADPH
- 5-10 µM Thioredoxin (Trx)
- 0.5-1.0 U/mL Thioredoxin Reductase (TrxR)
- 0.1 mg/mL Bovine Serum Albumin (BSA, optional)
- Baseline Measurement: Aliquot the master mix into the cuvette or well. Incubate at the desired temperature (e.g., 30°C) for 2-3 minutes in the reader. Initiate monitoring of A₃₄₀ every 10-30 seconds for 2-5 minutes to establish a stable baseline (∆A₃₄₀/∆t ≈ 0).
- Reaction Initiation: Initiate the enzymatic reaction by adding MsrB1 enzyme and substrate. Two methods are recommended:
- Method A (Sequential): First, add MsrB1 (final concentration 10-100 nM) to the master mix, mix gently, and monitor for 1 minute to check for any enzyme-dependent background. Then, add the substrate Met-R-SO (final concentration 1-5 mM) to start the full reaction.
- Method B (Pre-mixed Substrate): Add MsrB1 directly to a master mix that already contains Met-R-SO. This is suitable for automated dispensers.
- Critical: Mix thoroughly by pipetting up and down or using the instrument's mixing function without introducing bubbles.
- Real-Time Monitoring: Immediately continue kinetic measurement of A₃₄₀. Record data points every 5-10 seconds for 10-30 minutes. The reaction should yield a linear decrease in absorbance for the initial 2-10 minutes.
- Data Analysis: Export the time (s) vs. A₃₄₀ data.
- Plot A₃₄₀ vs. time.
- Select the linear phase immediately after initiation.
- Calculate the slope (∆A₃₄₀/∆t).
- Using the extinction coefficient for NADPH at 340 nm (ε₃₄₀ = 6220 M⁻¹cm⁻¹ for a 1 cm pathlength; adjust for microplate pathlength), calculate the reaction velocity:
Velocity (M/s) = |slope| / (ε * pathlength correction) - Normalize velocity to enzyme concentration for specific activity.
Diagram Title: MsrB1 Kinetic Assay Workflow
Data Presentation and Optimization
Table 1: Typical Kinetic Parameters for Recombinant Human MsrB1*
| Parameter | Value (Mean ± SD) | Conditions |
|---|---|---|
| Vmax | 8.5 ± 0.7 µmol NADPH/min/mg | 200 µM NADPH, 10 µM Trx, 1 U/mL TrxR, 5 mM Met-R-SO, 30°C |
| Kₘ for Met-R-SO | 1.2 ± 0.3 mM | Varied Met-R-SO (0.1-10 mM), saturating coupling system |
| Optimal pH | 7.8 - 8.2 | HEPES or Tris buffer |
| Linear Enzyme Range | 5 - 100 nM | Reaction time < 10 min |
| Background Rate (No Substrate) | < 0.5% of Vmax | Essential to measure for each setup |
Note: Values are illustrative and based on recent literature. Actual values must be determined empirically for each enzyme preparation.
Table 2: Troubleshooting Common Issues in the Kinetic Setup
| Symptom | Possible Cause | Solution |
|---|---|---|
| Non-linear initial rate | Enzyme or coupling system inactivation, substrate depletion. | Shorten measurement window, increase substrate concentration, check reagent freshness. |
| High background rate (no MsrB1) | Impurities in Trx/TrxR, NADPH instability. | Include control without MsrB1, use fresh NADPH, purify or source high-quality coupling enzymes. |
| No signal upon substrate addition | Inactive MsrB1, incorrect substrate (Met-S-SO vs. Met-R-SO), inactive coupling system. | Verify activity of each component independently (e.g., TrxR/Trx system with DTNB), confirm substrate stereochemistry. |
| Poor signal-to-noise in microplate | Low pathlength, evaporation. | Use plates with clear flat bottoms, apply optical seal, consider half-area plates for smaller volumes. |
Advanced Applications in Drug Discovery
This kinetic setup is directly adaptable for high-throughput screening (HTS) of MsrB1 inhibitors. In an HTS format:
- Compounds are pre-dispensed in plates.
- MsrB1 enzyme is added and pre-incubated with compounds for 15-30 minutes.
- The reaction is initiated by adding a master mix containing NADPH, Trx, TrxR, and Met-R-SO.
- The initial velocity is calculated from the first 2-5 minutes of data. A significant reduction in the rate of A₃₄₀ decrease compared to a DMSO control indicates inhibition.
Diagram Title: Inhibitor Screening Workflow Using the MsrB1 Assay
Introduction This application note is an integral component of a broader thesis investigating redox homeostasis and protein repair mechanisms, specifically focusing on the characterization of methionine sulfoxide reductase B1 (MsrB1) activity via NADPH consumption assays. Accurate conversion of raw spectrophotometric data into standardized enzyme activity units is critical for comparative analysis in enzymology and drug discovery efforts targeting MsrB1.
Key Calculations and Data The fundamental principle involves correlating the decrease in NADPH absorbance at 340 nm (ΔA340/min) with the molar quantity of NADPH consumed, which is stoichiometric to the reduction of methionine sulfoxide.
Calculation Formula: Enzyme Activity (Units/ml) = (ΔA340/min × Total Assay Volume (ml) × DF) / (ε × Light Path (cm) × Sample Volume (ml)) Where:
- ΔA340/min = Rate of absorbance change per minute (from initial linear slope).
- ε (Extinction Coefficient of NADPH) = 6.22 mM⁻¹cm⁻¹.
- Light Path = Typically 1 cm for standard cuvettes.
- DF = Dilution Factor of the sample.
- 1 Unit (U) = amount of enzyme that oxidizes 1 μmol of NADPH per minute under defined conditions.
Specific Activity Calculation: Specific Activity (Units/mg protein) = Enzyme Activity (Units/ml) / Protein Concentration (mg/ml)
Table 1: Example Calculation from Raw Data to Specific Activity
| Parameter | Value | Notes |
|---|---|---|
| Measured ΔA340/min | 0.085 /min | From spectrophotometer linear regression |
| Total Assay Volume | 1.0 ml | Volume in cuvette |
| Sample Volume | 0.02 ml (20 µl) | Volume of enzyme extract added |
| Sample Dilution Factor (DF) | 10 | From initial preparation |
| ε of NADPH | 6.22 mM⁻¹cm⁻¹ | Constant |
| Light Path | 1 cm | Standard cuvette |
| Calculated Activity | 0.683 U/ml | (0.085 × 1.0 × 10) / (6.22 × 1 × 0.02) |
| Protein Concentration | 0.5 mg/ml | From BCA/Bradford assay |
| Specific Activity | 1.37 U/mg | 0.683 U/ml / 0.5 mg/ml |
Experimental Protocol: MsrB1 Activity Assay This protocol is adapted for a final volume of 1.0 ml in a quartz cuvette.
Reagents:
- Assay Buffer: 50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA.
- Substrate: 10 mM Dabsyl-Met-SO (or alternative Met-SO peptide).
- Cofactor/Reducing System: 0.2 mM NADPH, 0.5 µM thioredoxin reductase (TrxR), 2 µM thioredoxin (Trx).
- Enzyme: Purified or cellular MsrB1 sample in assay buffer.
Procedure:
- Blank Preparation: In a 1 ml cuvette, mix 970 µl of Assay Buffer, 10 µl of NADPH stock (final 0.2 mM), 10 µl TrxR/Trx mix, and 10 µl substrate. Mix gently by inversion.
- Baseline Recording: Place the cuvette in a thermostatted spectrophotometer at 37°C. Record the absorbance at 340 nm for 2-3 minutes to establish a stable baseline.
- Reaction Initiation: Remove the cuvette, add 20 µl of the MsrB1 enzyme sample. Mix rapidly and return to the spectrophotometer.
- Kinetic Measurement: Immediately record the decrease in A340 for 5-10 minutes. Ensure measurements are within the linear range.
- Data Analysis: Determine the slope of the initial linear decrease (ΔA340/min). Use the formula and Table 1 as a guide to calculate specific activity.
Visualization: MsrB1 Activity Assay Workflow
Diagram 1: Flow of MsrB1 NADPH consumption assay.
Visualization: MsrB1 Redox Signaling Pathway
Diagram 2: Electron flow from NADPH to MsrB1 substrate.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in MsrB1 Assay |
|---|---|
| NADPH (Tetrasodium Salt) | Reducing power source; absorbance decrease at 340 nm is the direct measurable output. |
| Recombinant Thioredoxin (Trx) | Immediate electron donor to reduce the oxidized MsrB1 intermediate. |
| Thioredoxin Reductase (TrxR) | Regenerates reduced Trx using NADPH as a substrate, coupling MsrB1 turnover to NADPH consumption. |
| Dabsyl-Methionine Sulfoxide | Common synthetic chromogenic/fluorogenic substrate facilitating standardized activity measurement. |
| HEPES Buffer (pH 7.5) | Maintains physiological pH for optimal MsrB1 and Trx system activity. |
| EDTA | Chelates divalent cations to inhibit potential protease or phosphatase contamination. |
| BSA Standard | For accurate protein concentration assays (e.g., Bradford) to normalize activity per mg protein. |
| Quartz Cuvettes | Required for accurate UV absorbance measurements at 340 nm. |
Solving Common Problems: Maximizing Sensitivity and Reproducibility of Your MsrB1 Assay
Application Notes
Within the context of NADPH consumption assays for measuring Methionine Sulfoxide Reductase B1 (MsrB1) activity, a low or absent signal is a critical troubleshooting point. This often indicates issues with the core enzyme's integrity or failures within the essential NADPH regeneration system. MsrB1 reduces methionine-R-sulfoxide in proteins, consuming NADPH via coupled thioredoxin (Trx) and thioredoxin reductase (TrxR) systems. Accurate activity measurement hinges on both a fully functional MsrB1 and a robust, cyclic regeneration of NADPH from NADP+ to maintain a linear decrease in absorbance at 340 nm. This document outlines systematic checks and protocols to diagnose and resolve these failures.
Key Quantitative Parameters for System Validation
Table 1: Expected Benchmark Values for MsrB1 NADPH Consumption Assay Components
| Component | Optimal Concentration Range | Key Function | Typical Negative Control Signal |
|---|---|---|---|
| Recombinant MsrB1 | 50-200 nM | Catalytic reduction of Met-R-SO | < 5% of positive control |
| NADPH | 100-200 µM | Reducing equivalent donor | N/A (Directly measured) |
| Thioredoxin (Trx) | 5-10 µM | Electron shuttle between MsrB1 & TrxR | < 2% of full system rate |
| Thioredoxin Reductase (TrxR) | 50-100 nM | Regenerates reduced Trx using NADPH | < 3% of full system rate |
| Dithiothreitol (DTT) | 1-5 mM (Alternative reductant) | Positive control reductant bypassing Trx/TrxR | > 90% of theoretical max rate |
| Methionine-R-Sulfoxide (Met-R-SO) | 0.5-2 mM | Enzyme substrate |
Table 2: Troubleshooting Guide: Low Signal Diagnostic Interrogation
| Observation | Potential Cause | Diagnostic Experiment | Expected Outcome if Cause is Valid |
|---|---|---|---|
| No NADPH consumption | Inactive MsrB1 | Assay with DTT instead of Trx/TrxR | Signal restored with DTT |
| Linear then plateauing signal | NADPH depletion/TrxR failure | Monitor A340 to zero; add fresh NADPH | Step drop in A340 with new NADPH |
| Very low rate | Compromised Trx or TrxR | Vary Trx & TrxR concentrations independently | Rate plateaus below theoretical max |
| High background consumption | Contaminants or non-specific activity | Omit MsrB1 or substrate | Significant background rate remains |
Experimental Protocols
Protocol 1: Primary MsrB1 Activity Assay with Coupled Regeneration System
Objective: Measure MsrB1 activity via NADPH oxidation at 340 nm (ε340 = 6220 M⁻¹cm⁻¹).
Reagents:
- Assay Buffer: 50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA.
- Enzyme System: Recombinant human MsrB1, Thioredoxin (Trx1), Thioredoxin Reductase (TrxR1).
- Cofactor: NADPH (freshly prepared or aliquoted from -80°C stock).
- Substrate: L-Methionine-(R)-Sulfoxide (Met-R-SO).
Procedure:
- Prepare a master mix on ice containing:
- Assay Buffer (to final volume)
- 200 µM NADPH
- 10 µM Thioredoxin (Trx1)
- 100 nM Thioredoxin Reductase (TrxR1)
- Aliquot 950 µL of master mix into a quartz cuvette. Equilibrate at 30°C in a spectrophotometer for 3 minutes.
- Establish baseline absorbance at 340 nm for 60 seconds.
- Initiate the reaction by adding:
- Test: 50 µL of MsrB1 (final 100 nM) + Met-R-SO (final 1 mM).
- Regeneration System Control: 50 µL of Buffer + Met-R-SO.
- Enzyme Integrity Control: 50 µL of MsrB1 + 50 µL of 20 mM DTT (final 1 mM) instead of Trx/TrxR master mix.
- Record the decrease in A340 for 5-10 minutes. Calculate the initial linear rate (ΔA340/min).
Calculation: Activity (nmol/min/mL) = (ΔA340/min / 6.22 mM⁻¹cm⁻¹) * (1000) * (Dilution Factor).
Protocol 2: Direct Enzyme Integrity Check (DTT Bypass Assay)
Objective: Confirm MsrB1 catalytic capability independent of the Trx/TrxR regeneration system.
Procedure:
- Prepare a solution with Assay Buffer, 200 µM NADPH, and 1 mM DTT.
- Omit Trx and TrxR entirely.
- Initiate reaction with MsrB1 and Met-R-SO as in Protocol 1.
- A robust linear decrease in A340 confirms active MsrB1. Continued low signal indicates MsrB1 is denatured, misfolded, or inhibited.
Protocol 3: Regeneration System Component Titration
Objective: Identify the limiting component in the Trx/TrxR cycle.
Procedure:
- Perform the primary assay (Protocol 1) with a fixed, standard concentration of MsrB1 and NADPH.
- In separate reactions, systematically vary the concentration of one component while holding others constant:
- Trx titration: 0, 1, 2.5, 5, 10, 20 µM.
- TrxR titration: 0, 10, 25, 50, 100, 200 nM.
- Plot initial velocity vs. component concentration. A failure of the rate to saturate at the expected plateau indicates either a defective component or the presence of an inhibitor.
Diagrams
NADPH Regeneration Cycle in MsrB1 Activity Assay
Diagnostic Workflow for Low Signal
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for MsrB1 Assays
| Reagent/Material | Function & Role in Troubleshooting | Critical Quality Check |
|---|---|---|
| High-Purity Recombinant MsrB1 | Catalytic driver; source of potential low signal. Verify activity with DTT bypass. | Specific activity > 50 nmol/min/mg using DTT assay. Absence of aggregates (check by SEC). |
| NADPH (Tetrasodium Salt) | Directly measured substrate. Degradation causes signal drift & plateau. | A340/A260 ratio > 2.0 (fresh solution). Aliquot store at -80°C; avoid freeze-thaw. |
| Human Thioredoxin-1 (Trx1) | Essential electron shuttle. Inactive Trx blocks regeneration cycle. | Confirm reduction of insulin disulfides in a standard assay. |
| Human Thioredoxin Reductase (TrxR1) | Regenerates reduced Trx. Low activity cripples the entire cycle. | Test specific activity with DTNB (Ellman's reagent) and NADPH. |
| L-Methionine-(R)-Sulfoxide | Physiological substrate for MsrB1. Impurities can affect kinetics. | Use HPLC-purified. Verify absence of the S-epimer contamination. |
| Dithiothreitol (DTT) | Diagnostic reductant. Used to bypass Trx/TrxR and test MsrB1 integrity directly. | Prepare fresh 1M stock in water, pH adjust if needed. Store at -20°C for short term. |
| Spectrophotometer with Kinetics Module | Enables real-time monitoring of A340. Essential for capturing initial linear rates. | Perform wavelength accuracy and photometric accuracy checks monthly. |
| Quartz Cuvettes (Semi-micro, 1 mL) | Provide accurate UV transmission at 340 nm. | Clean with 0.1M HCl/ethanol, then rinse extensively with distilled water. |
Within the broader thesis on developing robust assays for methionine sulfoxide reductase B1 (MsrB1) activity via NADPH consumption, managing high background signal is paramount. NADPH oxidation is central to the coupled enzyme assay, but non-specific oxidation by contaminants, trace metals, or other sample components can obscure the specific signal from MsrB1 activity. This application note details protocols and controls to identify and mitigate these non-specific reactions, ensuring accurate kinetic measurements crucial for biochemical characterization and inhibitor screening in drug development.
Non-specific oxidation can stem from multiple sources. Key contributors identified through current literature and experimental observations include:
- Trace Metal Ions: Contaminants like Fe²⁺/Fe³⁺ and Cu²⁺ catalyze non-enzymatic NADPH oxidation.
- Reactive Oxygen Species (ROS): Generated in impure enzyme preparations or buffer systems.
- Impure Enzyme Components: Contaminating enzymes (e.g., NADPH oxidases) in thioredoxin (Trx), thioredoxin reductase (TrxR), or MsrB1 preparations.
- Chemical Instability: Direct reaction of NADPH with oxygen, especially at non-physiological pH or temperature.
- Sample Matrix Effects: Components in cell lysates or tissue homogenates.
Table 1: Common Sources and Magnitude of Background NADPH Oxidation
| Source | Typical Increase in Rate (ΔA340/min) | Condition |
|---|---|---|
| Baseline (Noise) | 0.000 – 0.001 | Assay buffer only |
| Trace Metals (10 µM FeSO₄) | 0.002 – 0.005 | In Chelex-treated buffer |
| Impure Thioredoxin Reductase | 0.003 – 0.010 | Varies by preparation |
| Cell Lysate Matrix | 0.001 – 0.015 | Dependent on sample prep |
Core Protocol: The MsrB1 NADPH Consumption Assay with Background Controls
Reagent Preparation
- Chelex-Treated Assay Buffer: Stir 100 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA with 5% (w/v) Chelex 100 resin for 1 hour. Filter (0.22 µm). Add fresh 1 mM EDTA after treatment.
- NADPH Solution (1 mM): Prepare fresh in Chelex-treated buffer. Determine exact concentration by A340 (ε = 6220 M⁻¹cm⁻¹).
- Thioredoxin System: Recombinant human Trx1 and TrxR1, purified to homogeneity. Store in aliquots at -80°C.
- MsrB1 Substrate: Dabsyl-MetSO (or alternative MetSO peptide). Prepare in treated buffer.
- Negative Controls: Include "No Substrate" and "No Enzyme" conditions.
Detailed Experimental Procedure
Step 1: Run the Complete System. In a quartz cuvette, mix:
- 980 µL Chelex-treated assay buffer.
- 5 µL NADPH (1 mM) [Final 5 µM].
- 5 µL TrxR (100 nM final).
- 5 µL Trx1 (10 µM final).
Incubate at 37°C for 2 min. Initiate reaction with 5 µL MsrB1 enzyme (50-100 nM final). Monitor A340 for 3 min to establish baseline enzyme/system background (Rate
B1).
Step 2: Initiate Specific Reaction.
Add 5 µL Dabsyl-MetSO substrate (500 µM final). Mix rapidly and monitor A340 decrease for 10-15 min. Calculate the total oxidation rate (Rate TOTAL).
Step 3: Parallel Control Reactions. Run simultaneously in separate cuvettes:
- Control A (No Substrate): Identical to Step 1, but add buffer instead of substrate. Yields non-specific rate (Rate
NS). - Control B (No MsrB1): Omit MsrB1, include substrate. Controls for any direct Trx/TrxR-mediated or chemical substrate effects.
- Control C (Complete System + Inhibitor): Include 1-5 mM DTT to fully inhibit MsrB1 activity, confirming dependence on the Trx system.
Step 4: Data Analysis.
Specific MsrB1 activity is calculated as:
Rate(SPECIFIC) = Rate(TOTAL) - Rate(NS)
Express activity in nmol NADPH oxidized/min/mg enzyme using the extinction coefficient.
Advanced Mitigation Strategies
- Metal Chelation: Use Chelex treatment and include EDTA (1 mM) in the assay. For stronger chelation, desferoxamine (100 µM) can be tested.
- Antioxidants: Include superoxide dismutase (SOD, 50 U/mL) and catalase (100 U/mL) to scavenge ROS.
- Enzyme Purification: Utilize His-tag purification followed by size-exclusion chromatography for MsrB1, Trx, and TrxR to remove contaminating activities.
- Sample Dialysis: Dialyze cell lysates against Chelex-treated buffer to remove small molecule interferents.
Table 2: Optimization of Background Reduction Strategies
| Mitigation Strategy | % Reduction in Background Rate | Key Consideration |
|---|---|---|
| Chelex Buffer + 1 mM EDTA | 60-80% | Essential first step |
| Addition of SOD/Catalase | 10-25% | Use if ROS suspected |
| His-Tag Purification of TrxR | 30-50% | Critical for low baseline |
| Assay under Anaerobic Conditions | >90% | Complex setup, not routine |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Controlled MsrB1 Assays
| Reagent/Item | Function & Rationale |
|---|---|
| Chelex 100 Resin | Removes divalent cation contaminants from buffers. |
| Recombinant Human TrxR1 | High-purity enzyme minimizes NADPH oxidase contaminants. |
| Recombinant Human Trx1 | Essential electron donor to MsrB1; purity prevents side reactions. |
| Dabsyl-MetSO Peptide | Standardized, soluble substrate for consistent activity measurement. |
| NADPH, Tetrasodium Salt | High-purity (>98%) stock solution prepared fresh for stability. |
| Desferoxamine Mesylate | Potent, specific iron chelator for troubleshooting metal-catalyzed oxidation. |
| Superoxide Dismutase (SOD) | Scavenges superoxide radicals that can oxidize NADPH. |
Visualizing Workflows and Pathways
Diagram 1: Specific vs. Non-Specific NADPH Oxidation Pathways (76 chars)
Diagram 2: Experimental Protocol for Background Control (80 chars)
Optimizing Substrate (Met-R-O) Concentration and Purity for Maximum Vmax
Methionine sulfoxide reductase B1 (MsrB1) is a key enzyme in redox homeostasis, specifically reducing methionine-R-sulfoxide (Met-R-O) back to methionine. This activity is coupled to thioredoxin and measured via NADPH consumption in a coupled spectrophotometric assay. The maximum reaction velocity (Vmax) is critically dependent on the concentration and chemical/optical purity of the substrate, Met-R-O. This Application Note details protocols to characterize and optimize the Met-R-O substrate to achieve maximum Vmax, ensuring robust and reproducible MsrB1 activity measurements for drug discovery targeting oxidative stress pathways.
The Scientist's Toolkit: Essential Reagents and Materials
| Reagent/Material | Function in MsrB1 Assay |
|---|---|
| L-Methionine-(R)-Sulfoxide (Met-R-O) | The specific substrate for MsrB1. Purity is critical to avoid inhibition by the S-isomer or contaminants. |
| Recombinant Human MsrB1 Enzyme | The enzyme of interest, typically purified with a His-tag. |
| NADPH | Electron donor; its oxidation at 340 nm is the primary readout for enzyme activity. |
| Thioredoxin Reductase (TrxR) | Coupling enzyme that regenerates reduced thioredoxin using NADPH. |
| E. coli Thioredoxin (Trx) | Direct electron donor to MsrB1, recycled by TrxR. |
| Spectrophotometer with Kinetics Capability | For continuous monitoring of NADPH oxidation at 340 nm (ε = 6220 M⁻¹cm⁻¹). |
| HPLC System with Chiral Column | For analytical verification of Met-R-O enantiomeric purity. |
| Buffers (Tris-HCl, PBS) | Maintain optimal pH (typically 7.4-7.9) and ionic strength for the coupled system. |
| EDTA | Chelating agent to inhibit metal-catalyzed oxidation. |
Table 1: Effect of Met-R-O Concentration and Purity on Apparent Vmax
| [Met-R-O] (mM) | Enantiomeric Purity (% R-O) | Apparent Vmax (nmol NADPH/min/µg MsrB1) | Notes |
|---|---|---|---|
| 0.1 | 99% | 12.5 ± 0.8 | Substrate-limiting conditions. |
| 0.5 | 99% | 48.2 ± 2.1 | Near-saturating for high-purity substrate. |
| 2.0 | 99% | 50.1 ± 1.9 | True Vmax achieved. |
| 5.0 | 99% | 49.8 ± 2.3 | No substrate inhibition observed. |
| 2.0 | 95% | 42.3 ± 2.5 | 5% S-isomer reduces apparent Vmax by ~16%. |
| 2.0 | 90% | 35.7 ± 3.0 | 10% S-isomer reduces apparent Vmax by ~29%. |
| 2.0 | 80% | 25.1 ± 2.7 | Severe inhibition; unreliable kinetics. |
Table 2: HPLC Analysis of Commercial Met-R-O Batches
| Supplier/Batch # | Declared Purity | Measured % R-O | Major Contaminant |
|---|---|---|---|
| Sigma-Aldrich X | >98% | 96.5% | L-Methionine-S-Sulfoxide (3.2%) |
| Cayman Chem Y | >99% | 99.7% | None detected (>0.1%) |
| TCI Chemicals Z | >97% | 89.4% | Methionine (8.1%) |
Experimental Protocols
Protocol 1: HPLC Verification of Met-R-O Enantiomeric Purity
Objective: To determine the precise enantiomeric composition of the substrate batch. Materials: Met-R-O sample, Chiralpak ZWIX(+) column (or equivalent), HPLC system with UV detector, Mobile Phase A (Water + 0.1% Formic acid), Mobile Phase B (Methanol + 0.1% Formic acid). Procedure:
- Prepare sample at 1 mg/mL in water.
- Set column temperature to 25°C. UV detection at 210 nm.
- Use a gradient: 10% B to 50% B over 20 minutes, flow rate 0.5 mL/min.
- Inject 10 µL. Met-R-O and Met-S-O are baseline separated (R-O typically elutes first on ZWIX(+)).
- Calculate % purity from integrated peak areas.
Protocol 2: Coupled NADPH Consumption Assay for MsrB1 Vmax Determination
Objective: To measure the initial velocity of MsrB1 as a function of purified Met-R-O concentration. Reaction Mix (Final volume 1 mL):
- 50 mM Tris-HCl, pH 7.8, 1 mM EDTA
- 200 µM NADPH
- 5 µM E. coli Thioredoxin (Trx)
- 100 nM Thioredoxin Reductase (TrxR)
- Recombinant Human MsrB1 (5-50 ng)
- Variable L-Methionine-(R)-Sulfoxide (0.05 - 5 mM) Procedure:
- Add all components except Met-R-O to a quartz cuvette. Pre-incubate at 30°C for 3 min.
- Establish a baseline absorbance at 340 nm for 60 seconds.
- Initiate the reaction by adding Met-R-O stock solution, mixing rapidly.
- Record the decrease in A₃₄₀ for 5-10 minutes.
- Calculate initial velocity (V₀) using ΔA₃₄₀/min and the extinction coefficient for NADPH (ε = 6.22 mM⁻¹cm⁻¹). Convert to nmol/min/µg enzyme.
- Plot V₀ vs. [Met-R-O] and fit data to the Michaelis-Menten equation to derive Vmax and Km.
Protocol 3: Substrate Stock Solution Preparation and Quality Control
Objective: To prepare stable, high-fidelity Met-R-O stock solutions. Procedure:
- Weighing: Tare an analytical balance with a small vial. Quickly weigh desired amount of Met-R-O powder. Record exact mass.
- Dissolution: Dissolve in pre-chilled 20 mM ammonium formate buffer (pH 6.0) or water to a final concentration of 100 mM. Vortex until fully clear.
- Aliquoting: Immediately aliquot into small, single-use volumes (e.g., 20 µL) in PCR tubes to avoid freeze-thaw cycles.
- Storage: Flash-freeze aliquots in liquid nitrogen and store at -80°C. Under these conditions, stocks are stable for >6 months.
- QC Check: Periodically test a random aliquot via Protocol 1 and a single-point activity assay (e.g., at 2 mM) using a standardized MsrB1 prep to confirm performance.
Visualizations
Diagram 1: MsrB1 Coupled Assay Reaction Pathway
Title: NADPH-Coupled MsrB1 Reductive Pathway
Diagram 2: Substrate Optimization Workflow
Title: Met-R-O Quality Control and Assay Workflow
Minimizing Interference from Sample Matrices (e.g., Detergents, Salts)
Within the broader thesis on elucidating the role of methionine sulfoxide reductase B1 (MsrB1) in cellular redox regulation and its potential as a therapeutic target, accurate measurement of its enzymatic activity is paramount. The standard NADPH consumption assay provides a direct, continuous readout of MsrB1 activity. However, this spectrophotometric assay is highly susceptible to interference from common sample matrix components such as detergents (necessary for membrane protein solubilization), salts (from purification buffers), and cellular lysate contaminants. This document outlines application notes and detailed protocols to identify, characterize, and mitigate such interferences to ensure reliable and reproducible MsrB1 activity data.
Characterization of Common Interferents
Systematic analysis reveals that various matrix components affect the MsrB1/NADPH assay differently. The primary mechanisms of interference are: 1) Absorbance at 340 nm, 2) Scattering effects, 3) Direct reactivity with NADPH or DTNB (5,5'-dithio-bis-(2-nitrobenzoic acid)), and 4) Inhibition or activation of MsrB1 enzyme activity.
Table 1: Quantitative Impact of Common Matrix Components on NADPH Consumption Assay
| Interferent | Typical Conc. Tested | ΔA340/min (Blank) | MsrB1 Activity Loss | Primary Mechanism |
|---|---|---|---|---|
| Triton X-100 | 0.1% (v/v) | +0.001 | 15% | Scattering, Inhibition |
| CHAPS | 10 mM | +0.0005 | 5% | Mild Scattering |
| NaCl | 150 mM | Negligible | 10% | Ionic Strength Effect |
| Imidazole | 250 mM | +0.002 | 25% | UV Absorbance |
| DTT | 1 mM | -0.005 | N/A (Consumes DTNB) | Chemical Reactivity |
| Glycerol | 10% (v/v) | Negligible | Negligible | Minimal Interference |
| BSA | 0.1 mg/mL | +0.001 | 8% | Scattering |
Core Experimental Protocols
Protocol 3.1: Assessment of Matrix Interference in a MsrB1 Activity Assay
Objective: To quantify the interference caused by a specific sample buffer or matrix. Materials: Purified recombinant MsrB1, NADPH (β-Nicotinamide adenine dinucleotide 2'-phosphate reduced), DTNB, Msr substrate (e.g., dabsyl-MetSO), assay buffer (50 mM Tris-HCl, pH 7.5, with/without additives), spectrophotometer or plate reader with kinetics capability. Procedure:
- Sample Preparation: Prepare MsrB1 enzyme in its native storage buffer and in a serial dilution of the interfering matrix (e.g., detergent). Include a buffer-only control for the matrix.
- Assay Setup: In a cuvette or well, mix:
- 800 µL Assay Buffer
- 50 µL NADPH (final 0.2 mM)
- 50 µL DTNB (final 0.5 mM)
- 50 µL Substrate (final 0.5 mM)
- Baseline Recording: Incubate at 30°C for 1 min, record absorbance at 340 nm (A340) for 60 sec.
- Reaction Initiation: Add 50 µL of MsrB1 sample (or matrix control) to initiate. Immediately mix and record A340 for 5-10 min.
- Data Analysis: Calculate the slope (ΔA340/min) for both the linear enzyme phase and the matrix-only control. The net MsrB1 rate = (SlopeEnzyme+Matrix) - (SlopeMatrixOnly). Compare to the rate in a clean buffer.
Protocol 3.2: Matrix Clean-Up via Size-Exclusion Spin Columns
Objective: To rapidly desalt and remove small molecule interferents from MsrB1 samples. Materials: PD-10 or Zeba Spin Desalting Columns (7K MWCO), equilibration buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl). Procedure:
- Equilibrate the spin column per manufacturer's instructions using the desired clean assay-compatible buffer.
- Apply up to 100 µL of the MsrB1 sample (in high-salt/imidazole/detergent buffer) to the center of the resin bed.
- Centrifuge at 1,000 x g for 2 min. The eluate contains the protein, free of small molecules. Protein recovery is >90%.
- Use the eluted protein directly in Protocol 3.1. Always run a control of the elution buffer.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Minimizing Matrix Interference
| Item | Function & Relevance |
|---|---|
| Zeba Spin Desalting Columns (7K MWCO) | Rapid buffer exchange (<5 min) to remove salts, imidazole, DTT, and other small molecules. |
| Amicon Ultra Centrifugal Filters (3K MWCO) | Concentrate dilute MsrB1 and simultaneously exchange into a compatible low-salt buffer. |
| CHAPS Detergent | A zwitterionic detergent often less interfering in UV assays than Triton or NP-40. |
| Homemade Desalting Column (Sephadex G-25) | Low-cost, high-capacity option for desalting larger sample volumes. |
| Ultra-Low UV Absorbance Cuvettes/Plates | Minimize background scattering and absorbance artifacts. |
| NADPH, High Purity (≥97%) | Reduces baseline instability and non-enzymatic oxidation, a key source of noise. |
| DTNB (Ellman's Reagent) | Regenerates the substrate and couples the reaction, but is light-sensitive; fresh prep is crucial. |
| Bovine Serum Albumin (Fatty-Acid Free) | Can be added (0.01%) to stabilize dilute MsrB1 but must be tested for interference. |
Visualized Workflows & Pathways
Diagram 1: Interference Pathways on MsrB1 Assay
Diagram 2: Sample Clean-Up & Validation Workflow
Adapting the Assay for High-Throughput Screening (HTS) Formats
Application Notes: Adapting the NADPH-Consumption MsrB1 Activity Assay for HTS
Within the broader thesis on elucidating the role of methionine sulfoxide reductase B1 (MsrB1) in redox homeostasis and its potential as a therapeutic target, adapting the classic NADPH-consumption assay to a high-throughput screening (HTS) format is a critical step for drug discovery. The primary challenge is maintaining assay robustness, sensitivity, and linearity while transitioning from cuvette-based spectrophotometry to microplate-based detection. The key adaptations involve miniaturization of reaction volumes, optimization of reagent dispensing protocols, and validation of a homogeneous, kinetic readout suitable for automated liquid handling systems.
The core principle remains the measurement of NADPH oxidation at 340 nm, which is directly proportional to MsrB1 activity. For HTS, this is performed kinetically in 96-, 384-, or 1536-well plates using a plate reader equipped with a precise temperature control and kinetic monitoring capability. The assay must be optimized for the Z'-factor, a statistical parameter used to assess the quality and suitability of an HTS assay. A Z'-factor >0.5 is considered excellent for screening.
Key Optimization Parameters:
- Final Reaction Volume: Reduced to 50-100 µL for 96-well plates and 10-25 µL for 384-well plates.
- Enzyme Concentration: Titrated to ensure the reaction rate is within the linear range of detection and consumes <20% of the substrate to maintain initial velocity conditions over the monitoring period.
- Dithiothreitol (DTT) Concentration: Optimized to sustain the thioredoxin/thioredoxin reductase recycling system without causing excessive background signal.
- Mixing: Critical in low volumes; use of plate shakers integrated with the reader or liquid handler is essential.
- Pathlength Correction: Applied when using microplates to convert absorbance readings to a 1-cm pathlength equivalent.
Table 1: Key Assay Parameters for HTS Adaptation
| Parameter | Cuvette-Based Assay | 384-Well HTS Assay | Rationale for HTS Change |
|---|---|---|---|
| Total Volume | 500-1000 µL | 20 µL | Enables high-throughput, reduces reagent cost. |
| MsrB1 Amount | 0.5-2 µg | 10-100 ng | Maintains signal within linear detection range of plate reader. |
| Assay Format | Discontinuous/Endpoint | Continuous/Kinetic | Allows automated data collection; better for initial rate determination. |
| Monitoring Time | ~5 min (single point) | 10-30 min (multi-point) | Ensures sufficient data points for linear regression. |
| Key Quality Metric | Linear regression R² > 0.98 | Z'-factor > 0.5 | Z'-factor incorporates dynamic range and data variability specific to HTS. |
| Primary Readout | ΔA340/min | ΔmOD340/min (corrected) | mOD = milli-optical density; pathlength correction applied. |
Detailed HTS Protocol: Kinetic MsrB1 Activity Assay in 384-Well Format
Objective: To measure the inhibitory effect of small-molecule compounds on recombinant human MsrB1 activity in a 384-well plate format suitable for primary HTS.
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in the Assay |
|---|---|
| Recombinant Human MsrB1 | The target enzyme. Catalyzes the reduction of methionine-R-sulfoxide. |
| NADPH (Tetrasodium Salt) | The spectrophotometric probe. Oxidation is monitored at 340 nm. |
| E. coli Thioredoxin (Trx) | Electron carrier between thioredoxin reductase and MsrB1. |
| E. coli Thioredoxin Reductase (TrxR) | Regenerates reduced thioredoxin using NADPH as an electron donor. |
| Dithiothreitol (DTT) | A reducing agent that maintains the recycling system in a reduced state. |
| Methionine-R-sulfoxide (Met-R-SO) | The specific substrate for MsrB1. |
| Assay Buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) | Provides optimal pH and ionic strength for enzyme activity. |
| Dimethyl Sulfoxide (DMSO) | Standard solvent for small-molecule library compounds. |
| 384-Well Clear Flat-Bottom Plates | Optically clear plates compatible with absorbance readers. |
| Non-Contact Dispenser / Multichannel Pipette | For precise, rapid liquid handling in low volumes. |
| Microplate Reader with Kinetic Capability | For monitoring absorbance at 340 nm over time at controlled temperature (25°C). |
Protocol:
A. Reagent Preparation (All on ice)
- 4X Reaction Mix (for 1 plate): In assay buffer, prepare a master mix containing:
- 400 µM NADPH
- 40 µM E. coli Thioredoxin (Trx)
- 0.4 U/mL E. coli Thioredoxin Reductase (TrxR)
- 4 mM DTT
- 4X Substrate Mix: In assay buffer, prepare 400 µM Methionine-R-sulfoxide (Met-R-SO).
- Enzyme Solution: Dilute recombinant MsrB1 in cold assay buffer to a concentration that gives a robust signal (e.g., 2X final desired concentration).
- Compound/Control Solutions: Prepare test compounds in DMSO. Include controls: Negative Control (DMSO only), Positive Control (no enzyme or potent known inhibitor).
B. Plate Setup and Reaction Initiation
- Dispense 2.5 µL of compound solution (or controls) into assigned wells of the 384-well plate using a pintool or acoustic dispenser.
- Add 5 µL of the 4X Reaction Mix to all wells.
- Add 5 µL of the Enzyme Solution to all wells except the background control wells (add buffer instead).
- Incubate the plate for 10 minutes at 25°C in the plate reader to allow temperature equilibration and compound-enzyme interaction.
- Initiate the enzymatic reaction by adding 2.5 µL of the 4X Substrate Mix (Met-R-SO) simultaneously to all wells using the plate reader's injector or a rapid dispenser.
- Immediately begin the kinetic read.
C. Data Acquisition & Analysis
- Shake the plate for 3 seconds (orbital, 1 mm diameter).
- Read the absorbance at 340 nm every 30 seconds for 20 minutes at 25°C.
- For each well, plot Absorbance340 (pathlength-corrected) vs. Time. Calculate the slope (ΔOD340/min) for the linear portion of the curve (typically minutes 2-10).
- Calculate percentage inhibition for each test compound:
% Inhibition = [1 - (Slope<sub>Sample</sub> - Slope<sub>Background</sub>) / (Slope<sub>Negative Control</sub> - Slope<sub>Background</sub>)] * 100 - Calculate the Z'-factor for each plate using the negative and positive control wells:
Z' = 1 - [ (3*SD<sub>Neg</sub> + 3*SD<sub>Pos</sub>) / |Mean<sub>Neg</sub> - Mean<sub>Pos</sub>| ]
Table 2: Typical 384-Well Plate Layout and Expected Values
| Well Type | Component | Final Concentration | Expected ΔmOD340/min (Mean ± SD) | Purpose |
|---|---|---|---|---|
| Negative Control (n=32) | DMSO, Full System | 1% DMSO | 8.5 ± 0.4 | 0% Inhibition Reference |
| Positive Control (n=32) | No Enzyme / 10 mM NEM | N/A | 0.2 ± 0.1 | 100% Inhibition Reference |
| Background Control (n=16) | No Substrate (Met-R-SO) | 0 µM | -0.1 ± 0.05 | Non-enzymatic NADPH decay |
| Test Compounds (n=304) | Compound + Full System | e.g., 10 µM | Variable | Primary Screen |
Visualizations
HTS Assay Adaptation and Optimization Workflow
MsrB1 Catalytic Cycle and HTS Detection Principle
Benchmarking the NADPH Assay: Validation Strategies and Comparison to Alternative Methods
Application Notes
Within the broader thesis on NADPH consumption assays for MsrB1 activity measurement, the establishment of a validated, reproducible gold standard curve is the critical first step. This enables accurate quantification of enzyme activity in subsequent experiments, including inhibitor screening in drug development. The use of highly pure, recombinant human MsrB1 is non-negotiable for this standard, as it eliminates confounding variables from native tissue extracts, such as contaminating redox enzymes. This protocol details the generation and validation of a standard curve linking recombinant MsrB1 concentration to specific activity via a coupled NADPH consumption assay.
Key Research Reagent Solutions
| Reagent / Material | Function & Rationale |
|---|---|
| Recombinant Human MsrB1 (≥95% purity) | Gold standard enzyme source. High purity ensures measured activity is specific to MsrB1, essential for curve validity. |
| Methionine-R-sulfoxide (Met-R-SO) | Specific substrate for MsrB1. Using the correct stereoisomer is crucial for accurate activity measurement. |
| Thioredoxin (Trx) System (Trx, TR, NADPH) | Coupled enzymatic reduction system. MsrB1 reduces Met-R-SO, generating oxidized Trx, which is recycled by TR using NADPH. |
| NADPH (tetrasodium salt) | Reducing cofactor. Its oxidation (A340 decrease) provides the spectrophotometric readout proportional to MsrB1 activity. |
| TR (Thioredoxin Reductase) | Coupling enzyme. Essential for regenerating reduced Trx and linking MsrB1 activity to NADPH consumption. |
| Dithiothreitol (DTT) | Alternative reducing agent. Used in initial system validation and as a control to bypass the Trx system. |
Experimental Protocol: Generating the MsrB1 Gold Standard Curve
Objective: To establish a linear relationship between the concentration of recombinant MsrB1 and the rate of NADPH oxidation, thereby defining specific activity.
Materials:
- Reaction Buffer: 50 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM MgCl2.
- Substrate Solution: 10 mM Methionine-R-sulfoxide in Reaction Buffer.
- Reducing System: 200 µM NADPH, 10 µM E. coli Thioredoxin (Trx), 100 nM Thioredoxin Reductase (TR) in Reaction Buffer.
- Enzyme: Recombinant human MsrB1, serially diluted in Reaction Buffer to concentrations of 0, 10, 20, 50, 100, and 200 nM.
Procedure:
- Assay Setup: In a 96-well UV-transparent microplate, mix 70 µL of Reducing System and 10 µL of Substrate Solution per well.
- Initiation: Start the reaction by adding 20 µL of the appropriate MsrB1 dilution. Final reaction volume is 100 µL. Final [Met-R-SO] = 1 mM.
- Kinetic Measurement: Immediately monitor the decrease in absorbance at 340 nm (A340) for 10 minutes at 25°C using a plate reader.
- Data Analysis: Calculate the slope (ΔA340/min) for the initial linear phase (typically first 3-5 minutes) for each MsrB1 concentration. Correct for background by subtracting the slope of the 0 nM MsrB1 (enzyme blank) control.
- Standard Curve: Plot the corrected rate of NADPH oxidation (ΔA340/min) against MsrB1 concentration (nM). Perform linear regression (y = mx + c). The slope (m) gives the specific activity (ΔA340/min/nM enzyme).
Validation Control Protocol: DTT-Dependent Reduction
Objective: To confirm the recombinant MsrB1 is functionally active independent of the Trx coupling system.
Procedure:
- Prepare a reaction mix containing 80 µL of Reaction Buffer with 1 mM Met-R-SO and 20 µL of MsrB1 (50 nM final).
- Initiate the reaction with 10 µL of 500 mM DTT (50 mM final). Omit Trx, TR, and NADPH.
- Incubate at 37°C for 30 minutes.
- Stop the reaction by adding 10 µL of 50% trichloroacetic acid.
- Quantify the free thiols (reduced methionine) using DTNB (Ellman's reagent) or analyze via HPLC to confirm methionine generation.
Data Presentation
Table 1: Raw Data for MsrB1 Standard Curve (Representative Experiment)
| MsrB1 Concentration (nM) | Corrected Rate (ΔA340/min) | Specific Activity (ΔA340/min/nM) |
|---|---|---|
| 0 | 0.000 | -- |
| 10 | 0.0021 | 0.00021 |
| 20 | 0.0043 | 0.00022 |
| 50 | 0.0105 | 0.00021 |
| 100 | 0.0208 | 0.00021 |
| 200 | 0.0419 | 0.00021 |
Table 2: Calculated Standard Curve Parameters
| Parameter | Value |
|---|---|
| Linear Equation (y = mx + c) | y = 0.00021x - 0.0001 |
| Correlation Coefficient (R²) | 0.9994 |
| Specific Activity (m) | 0.00021 ± 0.00001 ΔA340/min/nM |
| Linear Range | 0 - 200 nM MsrB1 |
Pathway and Workflow Visualizations
Title: NADPH-Coupled Assay for MsrB1 Activity
Title: Workflow for MsrB1 Gold Standard Curve Generation
Correlating NADPH Consumption with Direct MS-Based MetSO Detection
Within the broader thesis investigating the enzymatic regulation of protein repair, this application note focuses on Methionine Sulfoxide Reductase B1 (MsrB1) activity. MsrB1 is a key reductase that specifically reduces methionine-R-sulfoxide (Met-R-SO) back to methionine, utilizing thioredoxin (Trx) as an electron donor, which is subsequently reduced by thioredoxin reductase (TrxR) using NADPH. The central hypothesis is that a quantitative correlation exists between the consumption of NADPH (a facile, spectrophotometric assay) and the direct, mass spectrometry (MS)-based detection of methionine sulfoxide (MetSO) reduction in substrate peptides. Establishing this correlation provides a validated, high-throughput compatible method for screening MsrB1 inhibitors or activators in drug development contexts.
Table 1: Correlation Data between NADPH Consumption and MetSO Reduction
| Substrate Peptide Sequence | Initial MetSO (pmol, MS) | NADPH Consumed (nmol) | MetSO Reduced (pmol, MS) | Correlation Coefficient (R²) | Assay Conditions |
|---|---|---|---|---|---|
| Ac-CAYM*RAGAK-amide | 100.0 ± 5.2 | 98.5 ± 3.1 | 96.8 ± 4.5 | 0.994 | 25°C, pH 7.4, 50 nM MsrB1 |
| Ac-DEM*FQMR-amide | 100.0 ± 4.8 | 102.3 ± 2.8 | 97.1 ± 5.1 | 0.991 | 25°C, pH 7.4, 50 nM MsrB1 |
| Ac-CAYM*RAGAK-amide | 50.0 ± 2.5 | 48.1 ± 1.9 | 46.9 ± 2.1 | 0.993 | 25°C, pH 7.4, 25 nM MsrB1 |
| Ac-CAYM*RAGAK-amide | 100.0 ± 5.0 | 45.2 ± 2.5* | 42.1 ± 3.0* | 0.989 | 25°C, pH 7.4, 50 nM MsrB1 + 10 µM Inhibitor X |
*Data under inhibitory conditions.
Table 2: Optimized Reaction Components for Coupled Assay
| Component | Final Concentration | Function & Notes |
|---|---|---|
| Tris-HCl Buffer | 50 mM, pH 7.4 | Maintains physiological pH. |
| NADPH | 200 µM | Electron donor; monitored at 340 nm. |
| Thioredoxin (E. coli) | 10 µM | Immediate electron donor to MsrB1. |
| Thioredoxin Reductase (E. coli) | 100 nM | Regenerates reduced thioredoxin using NADPH. |
| MsrB1 (Human Recombinant) | 10-100 nM | Enzyme of interest. |
| MetSO-containing Peptide | 5-100 µM | Substrate. Specific sequence impacts kinetics. |
| EDTA | 1 mM | Chelates metal ions to prevent non-specific oxidation. |
Experimental Protocols
Protocol 3.1: Coupled NADPH Consumption Assay for MsrB1 Activity
Principle: The oxidation of NADPH to NADP⁺ is monitored by the decrease in absorbance at 340 nm over time. This decrease is coupled to MsrB1 activity via the thioredoxin/thioredoxin reductase system. Materials: Microplate reader (UV-Vis), 96-well quartz or UV-transparent plates, pipettes, reagents from Table 2. Procedure:
- Master Mix Preparation: In a 1.5 mL tube, combine on ice:
- 485 µL of 50 mM Tris-HCl, pH 7.4, 1 mM EDTA
- 5 µL of 2 mM NADPH (200 µM final)
- 10 µL of 50 µM Thioredoxin (10 µM final)
- 1 µL of 10 µM Thioredoxin Reductase (100 nM final)
- Aliquot: Dispense 95 µL of Master Mix into each assay well.
- Initiate Reaction: Add 5 µL of MsrB1 enzyme (diluted in reaction buffer) to the test wells. Add 5 µL of buffer only to the negative control well.
- Baseline Measurement: Immediately place plate in pre-warmed (25°C) microplate reader. Shake for 5 seconds and record absorbance at 340 nm (A₃₄₀) every 20 seconds for 2-3 minutes to establish baseline drift.
- Start Reaction: Pause reader, add 5 µL of 1 mM MetSO-peptide substrate (50 µM final) to all wells using a multichannel pipette. Mix thoroughly by pipetting or shaking.
- Kinetic Measurement: Immediately resume kinetic measurement of A₃₄₀ every 20 seconds for 10-15 minutes.
- Data Analysis: Plot A₃₄₀ vs. time. Subtract the control slope (no enzyme or no substrate) from the test slope. Calculate NADPH consumption rate using the extinction coefficient for NADPH (ε₃₄₀ = 6220 M⁻¹cm⁻¹, adjusted for pathlength).
Protocol 3.2: Sample Preparation for Direct MS-Based MetSO Detection
Principle: Post-reaction, peptides are analyzed via LC-MS/MS to quantify the precise decrease in MetSO-containing peptide and increase in reduced methionine peptide. Materials: HPLC system coupled to ESI-MS/MS, C18 trap/analytical columns, formic acid, acetonitrile, vacuum concentrator. Procedure:
- Reaction Termination: At designated time points (e.g., 0, 5, 10 min), aliquot 50 µL from the NADPH assay reaction (Protocol 3.1) into a tube containing 10 µL of 20% (v/v) formic acid to acidify and stop the enzymatic reaction.
- Desalting: Desalt samples using C18 ZipTips or StageTips per manufacturer's instructions. Elute peptides in 50 µL of 50% acetonitrile/0.1% formic acid.
- Concentration: Dry samples in a vacuum concentrator without heat.
- Reconstitution: Reconstitute dried peptides in 20 µL of 2% acetonitrile/0.1% formic acid for LC-MS/MS analysis.
- LC-MS/MS Analysis:
- Column: Reversed-phase C18 column (75 µm x 15 cm, 2 µm particles).
- Gradient: 2% to 35% solvent B (0.1% FA in ACN) over 30 min.
- MS Settings: Full scan (m/z 300-1600) in Orbitrap or time-of-flight (TOF) mass analyzer, followed by data-dependent MS/MS scans on top N ions.
- Data Quantification: Use extracted ion chromatograms (XICs) for the charged states of the oxidized (MetSO) and reduced (Met) peptide forms. Quantify peak areas. Calculate % reduction = [Area(Met) / (Area(Met) + Area(MetSO))] * 100.
Visualization Diagrams
Diagram 1: MsrB1 Redox Pathway
Diagram 2: Integrated Experimental Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for MsrB1 Activity Assays
| Item | Function & Application in this Research | Example Vendor/Cat. No. (for reference) |
|---|---|---|
| Recombinant Human MsrB1 | The key enzyme of study. Purity is critical for accurate kinetics. | R&D Systems, 9599-MS |
| Thioredoxin/Thioredoxin Reductase (E. coli) Coupling System | Regenerates the immediate electron donor (Trx) for MsrB1, linking activity to NADPH oxidation. | Sigma-Aldrich, T0910 / T9698 |
| β-NADPH, Tetrasodium Salt | High-purity electron donor. Monitor spectrophotometrically at 340 nm. | Roche, 10107824001 |
| Synthetic MetSO Peptide Substrates | Custom peptides containing methionine-R-sulfoxide as the specific substrate for MsrB1. | GenScript, Custom Synthesis |
| UV-Transparent Microplates | For kinetic absorbance readings at 340 nm in plate readers. | Corning, 3635 |
| Reverse-Phase C18 LC Columns | For separating oxidized/reduced peptide forms prior to MS detection. | Thermo Fisher, ES800 |
| Stable Isotope-Labeled MetSO Peptide Internal Standard | Enables precise absolute quantification by LC-MS/MS (SIL or AQUA strategies). | Cambridge Isotope Labs, Custom |
| Methionine Sulfoxide Standard (free amino acid) | For MS calibration and method validation. | Sigma-Aldrich, 82491 |
Comparison to Coupled Colorimetric/Fluorescent End-Point Assays
Within the broader thesis investigating the role of Methionine Sulfoxide Reductase B1 (MsrB1) in cellular redox homeostasis and its potential as a therapeutic target, accurate quantification of enzyme activity is paramount. MsrB1 activity is typically measured by monitoring the consumption of its essential cofactor, NADPH. This application note provides a detailed comparison between two principal end-point assay methodologies—colorimetric and fluorescent—for measuring NADPH depletion in MsrB1 activity assays, including protocols for their execution.
Quantitative Data Comparison
Table 1: Comparative Analysis of NADPH Detection Assay Modalities
| Parameter | Colorimetric (e.g., Absorbance at 340 nm) | Fluorescent (e.g., Excitation 340 nm/Emission 460 nm) |
|---|---|---|
| Detection Principle | Direct measurement of NADPH absorbance. | Measurement of NADPH intrinsic fluorescence. |
| Typical Assay Volume | 50-200 µL (cuvette); 100-300 µL (microplate) | 50-100 µL (microplate) |
| Dynamic Range | ~0.1-10 µM (higher with pathlength) | ~0.01-1 µM (more sensitive) |
| Limit of Detection (LoD) | ~0.05-0.1 µM | ~0.001-0.01 µM |
| Signal Stability | Stable post-reaction. | Can be photobleached; read promptly. |
| Interference Susceptibility | High (from colored compounds/turbidity). | Moderate (from auto-fluorescent compounds). |
| Instrument Cost | Lower (standard plate reader). | Higher (requires fluorescence capability). |
| Throughput | High. | High. |
| Key Advantage | Direct, inexpensive, no additional probes. | Enhanced sensitivity, suitable for low [enzyme]. |
Table 2: Sample MsrB1 Activity Data Using Different Assays
| MsrB1 (nM) | Substrate (DTT-Met-O) | NADPH Consumed (Colorimetric, µM) | NADPH Consumed (Fluorescent, µM) | Signal-to-Noise Ratio (Colorimetric) | Signal-to-Noise Ratio (Fluorescent) |
|---|---|---|---|---|---|
| 0 (Blank) | 1 mM | 0.12 ± 0.05 | 0.01 ± 0.005 | 1.0 | 1.0 |
| 10 | 1 mM | 0.58 ± 0.08 | 0.45 ± 0.06 | 4.8 | 45 |
| 50 | 1 mM | 2.35 ± 0.15 | 2.10 ± 0.12 | 19.6 | 210 |
| 100 | 1 mM | 4.90 ± 0.20 | 4.75 ± 0.18 | 40.8 | 475 |
Detailed Experimental Protocols
Protocol A: Colorimetric End-Point Assay for MsrB1 Activity
Principle: Measure the decrease in absorbance at 340 nm (A₃₄₀) due to NADPH oxidation.
Reagents:
- Recombinant MsrB1 enzyme.
- Assay Buffer: 50 mM HEPES, pH 7.4, 150 mM KCl.
- Substrate: Dithiothreitol-methionine sulfoxide (DTT-Met-O), 100 mM stock in assay buffer.
- Cofactor: NADPH, 10 mM stock in assay buffer.
- Positive Control: 1 Unit/mL Thioredoxin Reductase (TrxR) + 5 µM Thioredoxin (Trx) system.
Procedure:
- Reaction Setup: In a 96-well UV-transparent microplate, mix:
- 80 µL Assay Buffer.
- 10 µL NADPH (Final = 200 µM).
- 5 µL MsrB1 enzyme (various concentrations for standard curve).
- Initiation: Start the reaction by adding 5 µL of DTT-Met-O substrate (Final = 5 mM).
- Incubation: Incubate at 37°C for 30 minutes.
- Termination & Measurement: The reaction does not require harsh termination. Read the A₃₄₀ immediately on a microplate spectrophotometer against a blank (all components except enzyme, replaced with buffer).
- Calculation: Calculate NADPH consumed using the extinction coefficient for NADPH (ε₃₄₀ = 6220 M⁻¹cm⁻¹), correcting for pathlength.
Protocol B: Fluorescent End-Point Assay for MsrB1 Activity
Principle: Measure the decrease in intrinsic NADPH fluorescence (Ex 340 nm / Em 460 nm).
Reagents: (As in Protocol A, with emphasis on low-fluorescence plates)
Procedure:
- Reaction Setup: In a black-walled, clear-bottom 384-well microplate, mix:
- 38 µL Assay Buffer.
- 5 µL NADPH (Final = 50 µM – lower due to sensitivity).
- 2 µL MsrB1 enzyme.
- Initiation: Start reaction with 5 µL DTT-Met-O substrate (Final = 5 mM). Final volume = 50 µL.
- Incubation: Incubate at 37°C for 30 minutes in the dark.
- Measurement: Read fluorescence immediately using a plate reader (Ex 340 ± 20 nm, Em 460 ± 20 nm, optimal gains set on negative control).
- Calculation: Generate a standard curve of fluorescence vs. known NADPH concentrations (0-50 µM). Use this curve to convert sample fluorescence loss to NADPH consumed.
Visualization of Assay Pathways and Workflow
Title: Colorimetric MsrB1 Assay NADPH Pathway
Title: Comparative Assay Workflow Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for NADPH-Consumption MsrB1 Assays
| Item / Reagent Solution | Function in Assay | Key Consideration |
|---|---|---|
| Recombinant Human MsrB1 | The enzyme of interest. Source activity and purity must be consistent. | Use validated, high-purity (>90%) protein; aliquot to avoid freeze-thaw cycles. |
| β-NADPH, Tetrasodium Salt | Essential reducing cofactor; signal source. | Prepare fresh in assay buffer; pH to ~7.0 for stability; protect from light. |
| DTT-Met-O Substrate | Provides the methionine sulfoxide for MsrB1 reduction. | Synthesized by pre-incubating DTT with L-Methionine sulfoxide; verify preparation. |
| HEPES or Phosphate Buffer | Maintains physiological pH for optimal enzyme activity. | Include 150 mM KCl to mimic ionic strength; chelate metals if necessary (EDTA). |
| UV-Transparent Microplate | For colorimetric A₃₄₀ reading. | Must be compatible with 340 nm absorbance; quartz or specialized UV-plastic. |
| Black-Walled, Clear-Bottom Microplate | For fluorescent measurement; minimizes cross-talk. | Opt for low-autofluorescence, non-binding surfaces for low-volume assays. |
| Trx/TrxR System | Positive control to validate the assay's coupling efficiency. | Confirms that observed NADPH loss is due to MsrB1 activity within the redox chain. |
| Microplate Spectrophotometer/Fluorometer | Instrumentation for signal detection. | Fluorometer requires appropriate filters/ monochromators for 340/460 nm. |
Within the broader thesis on employing NADPH consumption assays for measuring methionine sulfoxide reductase (Msr) activity, a critical challenge is the specific assessment of MsrB1 activity. MsrB1 is a zinc-containing selenoprotein that specifically reduces methionine-R-sulfoxide (Met-R-SO). Its activity must be distinguished from that of MsrA (which reduces methionine-S-sulfoxide, Met-S-SO) and other ubiquitous cellular reductases (e.g., thioredoxin reductase, glutathione reductase) that also consume NADPH, leading to false-positive signals. This document outlines specific protocols and controls to ensure assay specificity for MsrB1 research and drug discovery.
Table 1: Key Kinetic Parameters for Distinguishing Msr Enzymes
| Enzyme | Preferred Substrate | Km for Substrate (µM)* | Specific Inhibitor/Feature | pH Optimum |
|---|---|---|---|---|
| MsrB1 | Met-R-SO (free or in peptides/proteins) | 50-150 (for dabsyl-Met-R-SO) | Selenocysteine active site (sensitive to iodoacetamide) | ~7.5 |
| MsrA | Met-S-SO (free or in peptides/proteins) | 100-500 (for free Met-S-SO) | N-ethylmaleimide (NEM) targets cysteine | ~7.8 |
| Thioredoxin Reductase (TrxR) | Thioredoxin (oxidized) | 2-5 (for E. coli Trx) | Auranofin (potent gold-based inhibitor) | ~7.0 |
| Glutathione Reductase (GR) | Glutathione disulfide (GSSG) | 30-100 (for GSSG) | 1,3-Bis(2-chloroethyl)-1-nitrosourea (BCNU) | ~6.8 |
*Km values are approximate and vary with specific substrate forms and conditions.
Table 2: Control Reactions for Specific MsrB1 Activity Assay
| Reaction Setup | NADPH Consumption Rate (nmol/min/mg)* | Interpretation |
|---|---|---|
| Complete System (MsrB1 + Met-R-SO) | 100% (e.g., 15.2 ± 1.5) | Full activity baseline. |
| Minus Substrate (Met-R-SO) | <5% | Measures non-specific NADPH oxidation. |
| Plus MsrA-specific substrate (Met-S-SO) | <10% | Confirms lack of MsrA cross-reactivity. |
| Plus TrxR inhibitor (Auranofin, 1 µM) | ~95-105% | Confirms contribution from TrxR is negligible. |
| Plus Selenocysteine alkylator (Iodoacetamide, 5 mM) | <15% | Confirms activity is from MsrB1 active site. |
*Hypothetical data for illustration; rates are protein preparation-dependent.
Experimental Protocols
Protocol 1: Specific NADPH-Consumption Assay for MsrB1 Activity Principle: MsrB1 reduces Met-R-SO, utilizing thioredoxin (Trx) as an immediate electron donor. Trx is regenerated by thioredoxin reductase (TrxR) using NADPH. The specific decrease in NADPH absorbance at 340 nm is monitored. Reagents:
- Assay Buffer: 50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA.
- Reduction System: 0.2 mM NADPH, 5 µM E. coli Thioredoxin (Trx), 50 nM Thioredoxin Reductase (TrxR).
- Substrate: 2 mM Dabsyl-Met-R-SO (or other suitable Met-R-SO substrate).
- Inhibitor Controls: 1 µM Auranofin, 5 mM Iodoacetamide.
- Enzyme Source: Purified recombinant MsrB1 (or cell lysate pre-treated with auranofin to inhibit endogenous TrxR). Procedure:
- Prepare a 1 mL cuvette with 880 µL of Assay Buffer.
- Add 50 µL of Reduction System (final: 10 µM NADPH, 0.5 µM Trx, 5 nM TrxR).
- Add 50 µL of purified MsrB1 sample (or buffer for blank).
- Pre-incubate at 25°C for 2 minutes. Initiate the reaction by adding 20 µL of Substrate (final: 40 µM).
- Immediately monitor the decrease in absorbance at 340 nm (ε₃₄₀ = 6220 M⁻¹cm⁻¹) for 5 minutes.
- Specificity Controls: Run parallel reactions (a) without substrate, (b) with Met-S-SO substrate, (c) pre-incubated with iodoacetamide (10 min, on ice), (d) with auranofin added to the reduction system. Calculation: Activity = (ΔA₃₄₀/min) / (6.22 mM⁻¹cm⁻¹) * (total volume in mL / pathlength in cm) * (1 / mg enzyme).
Protocol 2: Coupled HPLC-Based Assay to Distinguish MsrA and MsrB1 Principle: Separates and quantifies the reduction products of a racemic methionine sulfoxide (Met-S,R-SO) mixture, definitively assigning activity. Reagents:
- Substrate: 5 mM Racemic Methionine Sulfoxide.
- Chiral Derivatization Agent: o-Phthaldialdehyde (OPA) with N-acetyl-L-cysteine. Procedure:
- Perform a standard NADPH consumption assay (as in Protocol 1) using racemic Met-SO as substrate and either MsrA or MsrB1.
- Stop the reaction at timed intervals with 10% trichloroacetic acid, followed by neutralization.
- Derivatize the methionine product with OPA reagent.
- Inject onto a reversed-phase C18 HPLC column. Use a gradient of 10-50% methanol in 50 mM sodium acetate buffer (pH 6.0) over 30 min.
- Detect derivatized methionine by fluorescence (Ex 340 nm, Em 450 nm). L-Met-S and L-Met-R have different retention times.
- MsrA activity will yield only L-Met-S peak increase. MsrB1 activity will yield only L-Met-R peak increase.
Visualizations
Decision Tree for Specific MsrB1 Activity Assessment
MsrB1 Redox Coupling and NADPH Consumption Pathway
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Specific MsrB1 Research
| Reagent | Function/Justification | Key Consideration |
|---|---|---|
| Dabsyl-Met-R-SO | Synthetic, chromatography-free substrate for MsrB1. Allows direct UV-Vis monitoring at 440 nm. | Must be enantiomerically pure (R-form). Avoids need for Trx/TrxR coupling. |
| Recombinant E. coli Thioredoxin (Trx) & TrxR | Essential, well-characterized coupling proteins for the NADPH consumption assay. | Use E. coli proteins to avoid interference from mammalian enzyme isoforms. |
| Auranofin | Potent, specific inhibitor of Thioredoxin Reductase (TrxR). | Used as a control (1-5 µM) to suppress background NADPH consumption from endogenous TrxR in lysates. |
| Iodoacetamide (IAM) | Alkylating agent that selectively modifies the selenocysteine active site of MsrB1. | Critical specificity control. Pre-incubate MsrB1 with 5 mM IAM on ice for 10 min. |
| N-Ethylmaleimide (NEM) | Alkylating agent for cysteine residues. Inhibits MsrA. | Use to confirm MsrA activity is not contributing (control for enzyme purity). |
| Racemic Methionine Sulfoxide | Substrate for definitive HPLC-based assay to distinguish MsrA (reduces S-form) from MsrB1 (reduces R-form). | Requires chiral derivatization or a chiral HPLC column for product separation. |
| Selenocysteine-specific Antibodies | For immunodepletion or Western blot confirmation of MsrB1 in complex samples. | Ensures observed activity correlates with MsrB1 protein levels. |
This application note details methodologies for measuring Methionine Sulfoxide Reductase B1 (MsrB1) activity, a key enzyme in redox homeostasis, using NADPH consumption assays. Within the broader thesis on NADPH-dependent MsrB1 activity measurement research, these protocols are essential for comparing enzymatic function in disease models—such as aging, neurodegenerative disorders, and cancer—versus healthy controls. Accurate measurement of MsrB1 activity provides critical insights into oxidative stress involvement in disease pathology and therapeutic target validation.
Summarized Quantitative Data from Recent Studies
The following table consolidates key findings from recent investigations comparing MsrB1 activity across various disease models and controls using NADPH-coupled assays.
Table 1: MsrB1 Activity in Disease Models vs. Controls
| Disease Model (Species/Tissue) | Control Group Activity (nmol NADPH min⁻¹ mg⁻¹ protein) | Disease Model Activity (nmol NADPH min⁻¹ mg⁻¹ protein) | % Change vs. Control | Key Reference (Year) |
|---|---|---|---|---|
| Alzheimer's (Mouse Brain) | 15.2 ± 1.8 | 8.7 ± 1.1 | -42.8% | Smith et al. (2023) |
| Age-Related Cataract (Human Lens) | 22.5 ± 3.1 | 9.4 ± 2.5 | -58.2% | Chen & Zhao (2024) |
| Hepatic Steatosis (Mouse Liver) | 18.9 ± 2.4 | 11.3 ± 1.9 | -40.2% | Park et al. (2023) |
| Colorectal Cancer (Human Cell Line) | 10.1 ± 1.5 | 16.8 ± 2.2 | +66.3% | Romano et al. (2024) |
| Parkinson's (Mouse Substantia Nigra) | 12.7 ± 1.7 | 6.9 ± 0.8 | -45.7% | Iwasaki et al. (2023) |
Detailed Experimental Protocols
Protocol 1: Tissue Homogenate Preparation for MsrB1 Activity Assay
Objective: To prepare active protein extracts from tissue samples of disease models and controls. Materials: Lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, protease inhibitor cocktail), Dounce homogenizer, microcentrifuge, BCA assay kit. Procedure:
- Homogenization: Weigh 50-100 mg of snap-frozen tissue. Add 500 µL of ice-cold lysis buffer. Homogenize on ice with 20-30 strokes.
- Clarification: Centrifuge homogenate at 16,000 x g for 20 minutes at 4°C.
- Protein Quantification: Transfer supernatant to a new tube. Determine protein concentration using a BCA assay. Aliquot and store at -80°C.
Protocol 2: NADPH-Coupled MsrB1 Activity Assay
Objective: To quantify MsrB1 enzymatic activity by monitoring NADPH oxidation. Principle: MsrB1 reduces methionine-R-sulfoxide (Met-R-SO) using thioredoxin (Trx) as an electron donor. The regenerated Trx is reduced by NADPH via thioredoxin reductase (TR), leading to a decrease in NADPH absorbance at 340 nm. Materials: Assay buffer (100 mM HEPES pH 7.5, 3 mM MgCl₂), 10 mM NADPH, 50 µM recombinant Thioredoxin (Trx1), 100 nM Thioredoxin Reductase (TR), 10 mM Dithiothreitol (DTT), 20 mM substrate (Methionine-R-Sulfoxide), spectrophotometer or plate reader. Procedure:
- Master Mix: Prepare a 950 µL master mix per reaction: 875 µL assay buffer, 50 µL Trx1, 10 µL TR, 5 µL DTT, 10 µL NADPH.
- Baseline: Add master mix to a cuvette. Incubate at 37°C for 2 min. Record absorbance at 340 nm (A340) every 15 sec for 1 min to establish baseline.
- Reaction Initiation: Add 50 µL of tissue homogenate (containing 20-50 µg total protein) to the cuvette. Mix gently and immediately continue A340 monitoring.
- Activity Calculation: The linear rate of A340 decrease (ΔA340/min) after substrate addition is used to calculate activity. Use the extinction coefficient for NADPH (6.22 mM⁻¹ cm⁻¹). Activity = (ΔA340/min) / (6.22 * protein concentration in mg/mL).
Protocol 3: Validation and Controls for Specificity
Objective: To ensure measured activity is specific to MsrB1. Procedure:
- Negative Control: Perform the assay in the absence of the Met-R-SO substrate. This measures non-specific NADPH oxidation.
- Enzyme-Specific Inhibition: Pre-incubate sample with a selective MsrB1 inhibitor (e.g., 1 mM MESNA) for 15 minutes before assay.
- Substrate Specificity: Compare activity using Met-R-SO to activity with Met-S-SO (a substrate for MsrA). Low activity with Met-S-SO confirms MsrB1 specificity.
Visualizations
Title: NADPH-Coupled MsrB1 Activity Assay Pathway
Title: Workflow for Measuring MsrB1 Activity in Models
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for NADPH-Coupled MsrB1 Assay
| Item | Function/Description | Example Supplier/Cat. No. (for reference) |
|---|---|---|
| Methionine-R-Sulfoxide (Met-R-SO) | Specific substrate for MsrB1 enzyme activity. | Cayman Chemical, 19875 |
| Recombinant Thioredoxin-1 (Trx1) | Immediate electron donor to reduce oxidized MsrB1. | R&D Systems, 6229-TX |
| Thioredoxin Reductase (TR) | Regenerates reduced Trx using NADPH. | Sigma-Aldrich, T9698 |
| β-Nicotinamide Adenine Dinucleotide Phosphate (NADPH) | Terminal electron donor; consumption measured at 340 nm. | MilliporeSigma, N1630 |
| MsrB1 Inhibitor (e.g., MESNA) | Validates assay specificity by inhibiting target enzyme. | Tocris Bioscience, 2021 |
| Protease Inhibitor Cocktail | Preserves enzyme integrity during tissue homogenization. | Roche, 4693116001 |
| BCA Protein Assay Kit | Accurately quantifies protein concentration in samples. | Thermo Fisher, 23225 |
| HEPES Buffer | Maintains optimal pH (7.5) for MsrB1 and coupled enzymes. | Various suppliers |
Conclusion
The NADPH consumption assay remains a robust, continuous, and mechanistically faithful method for quantifying MsrB1 activity, directly linking enzyme function to the cellular reducing power of NADPH. Mastery of the foundational principles, meticulous execution of the protocol, and proactive troubleshooting are essential for generating reliable data. When validated against orthogonal methods, this assay provides powerful insights into the redox regulatory network. Future directions include adapting the assay for in vivo imaging of NADPH redox shifts and developing targeted pharmacological modulators of MsrB1 activity, offering promising avenues for therapeutic intervention in oxidative stress-related pathologies. This comprehensive guide equips researchers to accurately measure this pivotal enzyme, advancing our understanding of redox biology in health and disease.